

HDAC4-regulated STAT1 activation mediates platinum resistance in ovarian cancer
- PMID: 21571862
- PMCID: PMC3130134
- DOI: 10.1158/0008-5472.CAN-10-4111
HDAC4-regulated STAT1 activation mediates platinum resistance in ovarian cancer
Abstract
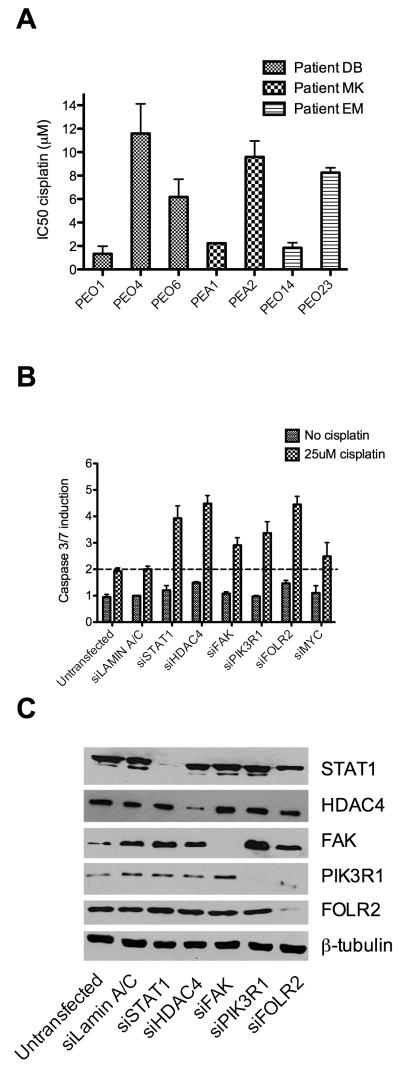
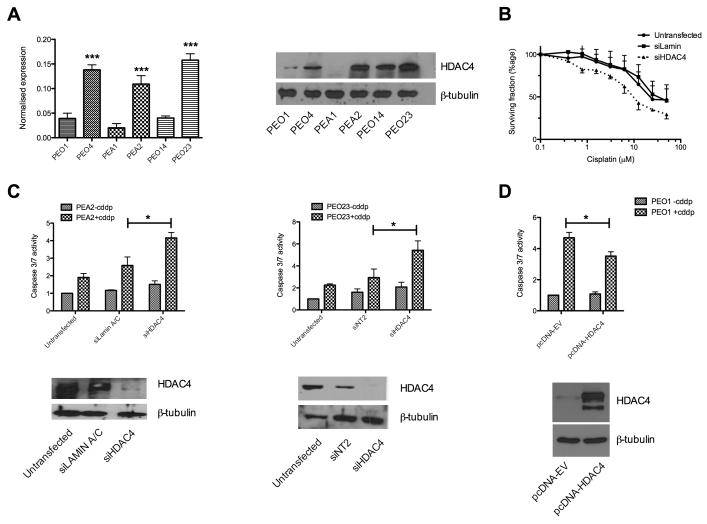
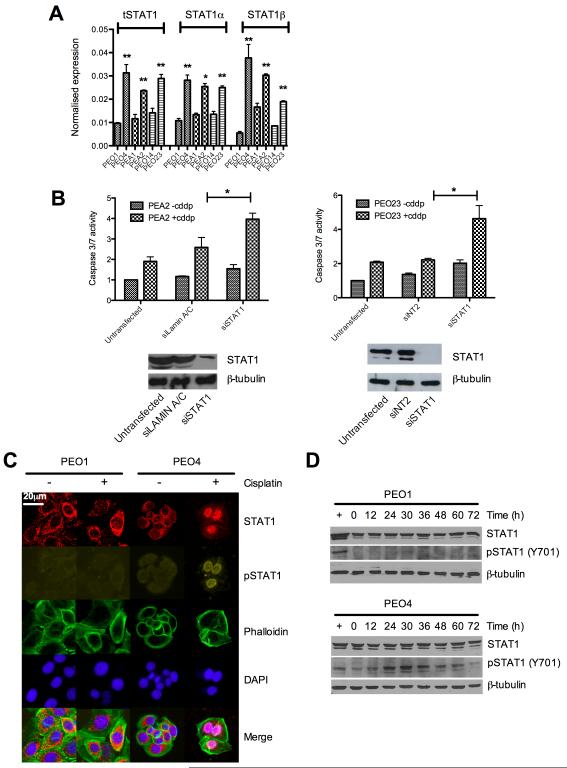
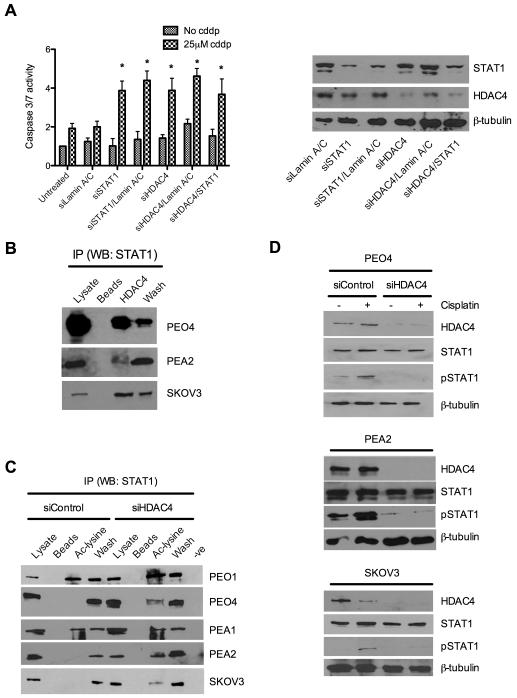
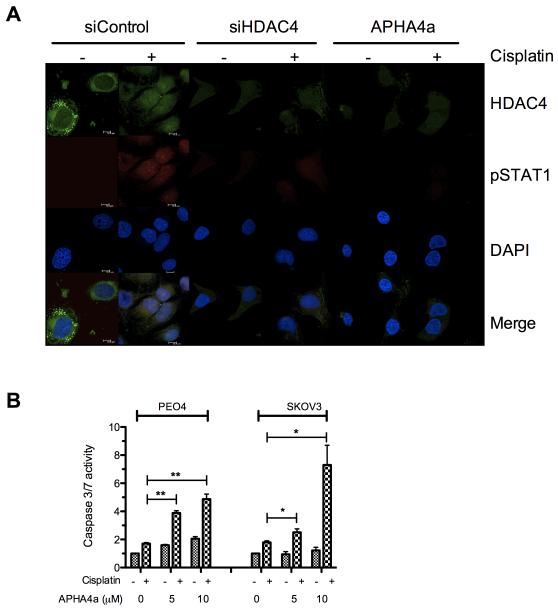
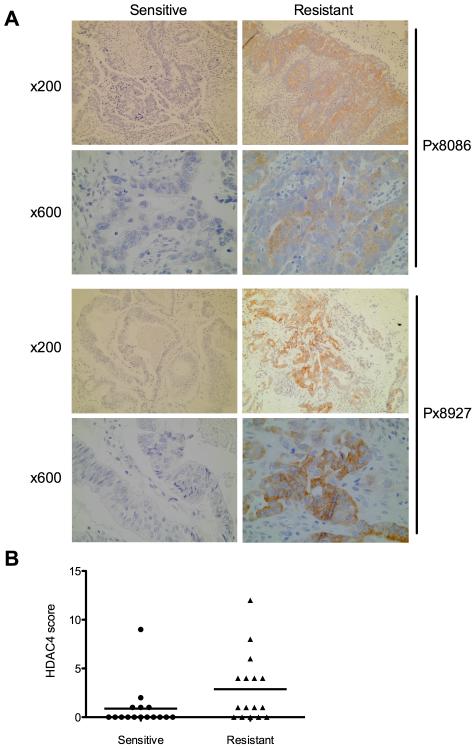
Ovarian cancer frequently acquires resistance to platinum chemotherapy, representing a major challenge for improving patient survival. Recent work suggests that resistant clones exist within a larger drug-sensitive cell population prior to chemotherapy, implying that resistance is selected for rather than generated by treatment. We sought to compare clinically derived, intrapatient paired models of initial platinum response and subsequent resistant relapse to define molecular determinants of evolved resistance. Transcriptional analysis of a matched cell line series from three patients with high-grade serous ovarian cancer before and after development of clinical platinum resistance (PEO1/PEO4/PEO6, PEA1/PEA2, PEO14/PEO23) identified 91 up- and 126 downregulated genes common to acquired resistance. Significantly enhanced apoptotic response to platinum treatment in resistant cells was observed following knockdown of histone deacetylase (HDAC) 4, FOLR2, PIK3R1, or STAT1 (P < 0.05). Interestingly, HDAC4 and STAT1 were found to physically interact. Acetyl-STAT1 was detected in platinum-sensitive cells but not in HDAC4 overexpressing platinum-resistant cells from the same patient. In resistant cells, STAT1 phosphorylation/nuclear translocation was seen following platinum exposure, whereas silencing of HDAC4 increased acetyl-STAT1 levels, prevented platinum-induced STAT1 activation, and restored cisplatin sensitivity. Conversely, matched sensitive cells were refractory to STAT1 phosphorylation on platinum treatment. Analysis of 16 paired tumor biopsies taken before and after development of clinical platinum resistance showed significantly increased HDAC4 expression in resistant tumors [n = 7 of 16 (44%); P = 0.04]. Therefore, clinical selection of HDAC4-overexpressing tumor cells upon exposure to chemotherapy promotes STAT1 deacetylation and cancer cell survival. Together, our findings identify HDAC4 as a novel, therapeutically tractable target to counter platinum resistance in ovarian cancer.
©2011 AACR.
Figures
References
-
- Herzog TJ. Recurrent ovarian cancer: how important is it to treat to disease progression? Clin Cancer Res. 2004;10:7439–49. - PubMed
-
- Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer. 2003;3:502–16. - PubMed
-
- Kartalou M, Essigmann JM. Mechanisms of resistance to cisplatin. Mutat Res. 2001;478:23–43. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
