

Click-generated triazole ureas as ultrapotent in vivo-active serine hydrolase inhibitors
- PMID: 21572424
- PMCID: PMC3118922
- DOI: 10.1038/nchembio.579
Click-generated triazole ureas as ultrapotent in vivo-active serine hydrolase inhibitors
Erratum in
- Nat Chem Biol. 2012 Mar;8(3):318
Abstract
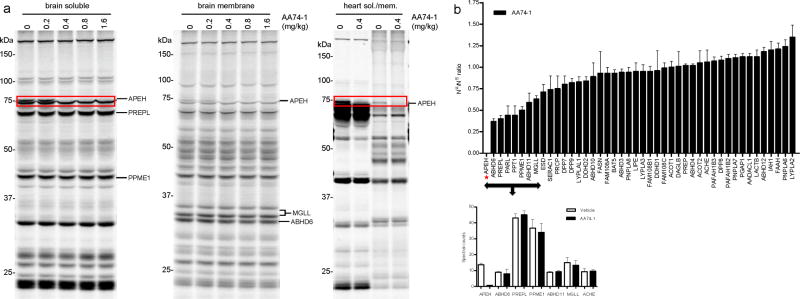
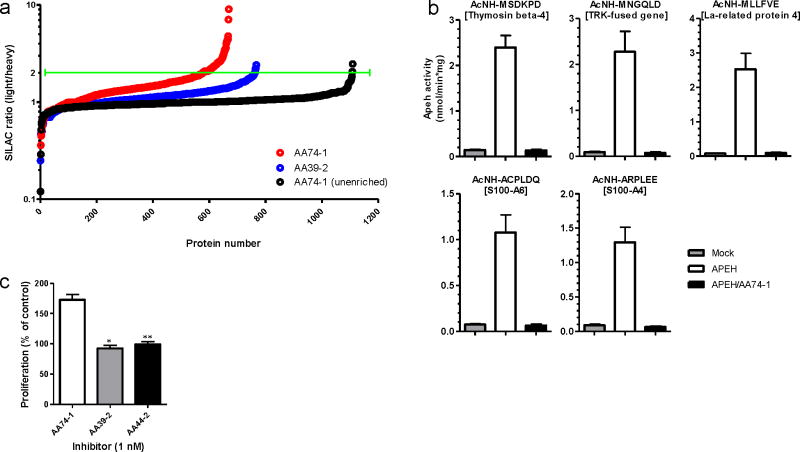
Serine hydrolases are a diverse enzyme class representing ∼1% of all human proteins. The biological functions of most serine hydrolases remain poorly characterized owing to a lack of selective inhibitors to probe their activity in living systems. Here we show that a substantial number of serine hydrolases can be irreversibly inactivated by 1,2,3-triazole ureas, which show negligible cross-reactivity with other protein classes. Rapid lead optimization by click chemistry-enabled synthesis and competitive activity-based profiling identified 1,2,3-triazole ureas that selectively inhibit enzymes from diverse branches of the serine hydrolase class, including peptidases (acyl-peptide hydrolase, or APEH), lipases (platelet-activating factor acetylhydrolase-2, or PAFAH2) and uncharacterized hydrolases (α,β-hydrolase-11, or ABHD11), with exceptional potency in cells (sub-nanomolar) and mice (<1 mg kg(-1)). We show that APEH inhibition leads to accumulation of N-acetylated proteins and promotes proliferation in T cells. These data indicate 1,2,3-triazole ureas are a pharmacologically privileged chemotype for serine hydrolase inhibition, combining broad activity across the serine hydrolase class with tunable selectivity for individual enzymes.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
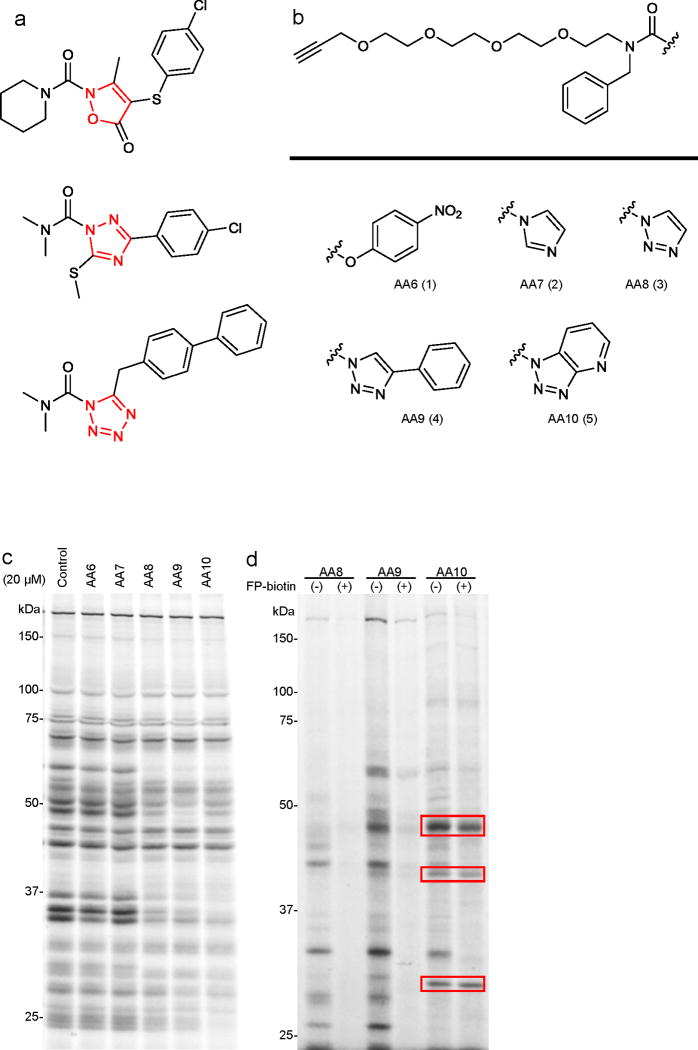
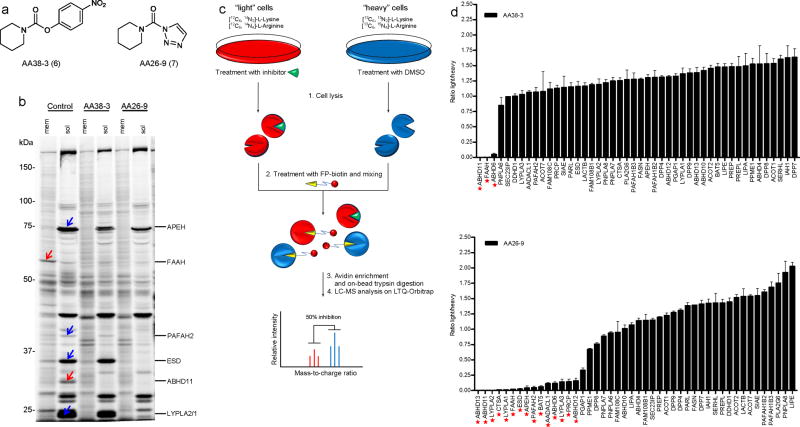
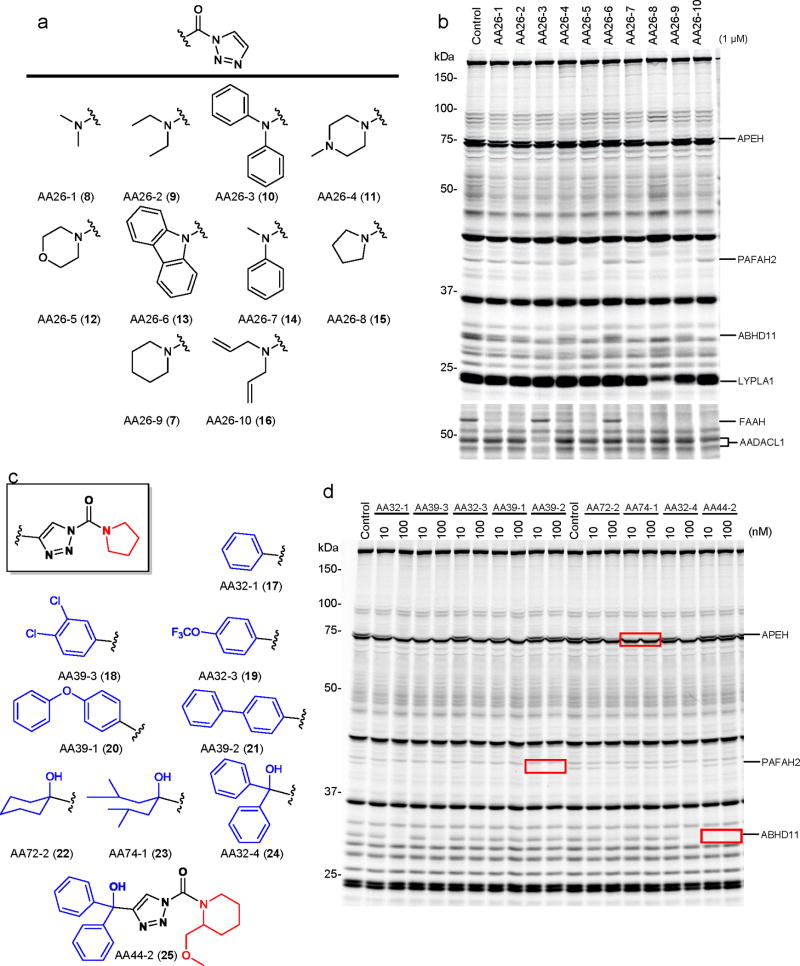
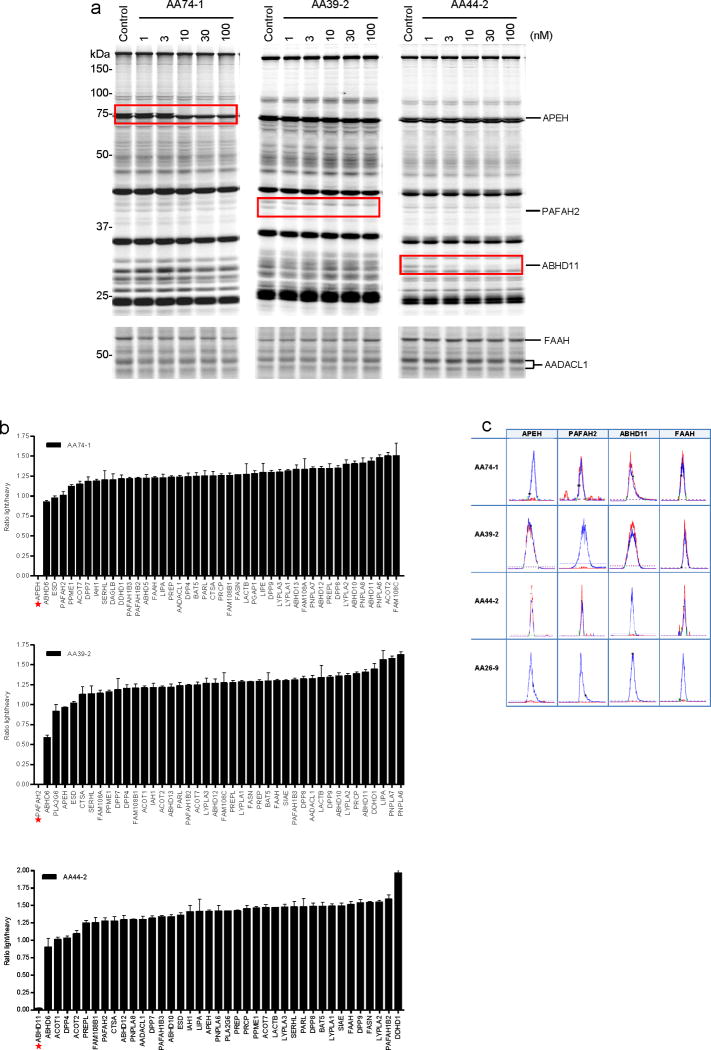
References
-
- Henness S, Perry CM. Orlistat: a review of its use in the management of obesity. Drugs. 2006;66:1625–56. - PubMed
-
- Thornberry NA, Weber AE. Discovery of JANUVIA (Sitagliptin), a selective dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. Curr Top Med Chem. 2007;7:557–68. - PubMed
-
- Kluge AF, Petter RC. Acylating drugs: redesigning natural covalent inhibitors. Curr Opin Chem Biol. 2010;14:421–7. - PubMed
-
- Birks J, Grimley Evans J, Iakovidou V, Tsolaki M, Holt FE. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst Rev. 2009:CD001191. - PubMed
Publication types
MeSH terms
Substances
Associated data
- PubChem-Substance/119526573
- PubChem-Substance/119526574
- PubChem-Substance/119526575
- PubChem-Substance/119526576
- PubChem-Substance/119526577
- PubChem-Substance/119526578
- PubChem-Substance/119526579
- PubChem-Substance/119526580
- PubChem-Substance/119526581
- PubChem-Substance/119526582
- PubChem-Substance/119526583
- PubChem-Substance/119526584
- PubChem-Substance/119526585
- PubChem-Substance/119526586
- PubChem-Substance/119526587
- PubChem-Substance/119526588
- PubChem-Substance/119526589
- PubChem-Substance/119526590
- PubChem-Substance/119526591
- PubChem-Substance/119526592
- PubChem-Substance/119526593
- PubChem-Substance/119526594
- PubChem-Substance/119526595
- PubChem-Substance/119526596
- PubChem-Substance/119526597
- PubChem-Substance/119526598
- PubChem-Substance/119526599
- PubChem-Substance/119526600
- PubChem-Substance/119526601
- PubChem-Substance/119526602
- PubChem-Substance/119526603
- PubChem-Substance/119526604
- PubChem-Substance/119526605
- PubChem-Substance/119526606
- PubChem-Substance/119526607
- PubChem-Substance/119526608
- PubChem-Substance/119526609
- PubChem-Substance/119526610
- PubChem-Substance/119526611
- PubChem-Substance/119526612
- PubChem-Substance/119526613
- PubChem-Substance/119526614
- PubChem-Substance/119526615
- PubChem-Substance/119526616
- PubChem-Substance/119526617
- PubChem-Substance/119526618
- PubChem-Substance/119526619
- PubChem-Substance/119526620
- PubChem-Substance/119526621
- PubChem-Substance/119526622
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
