

Alzheimer's disease
- PMID: 21576255
- PMCID: PMC3119915
- DOI: 10.1101/cshperspect.a004457
Alzheimer's disease
Abstract
Over the last three decades, advances in biochemical pathology and human genetics have illuminated one of the most enigmatic subjects in biomedicine--neurodegeneration. Eponymic diseases of the nervous system such as Alzheimer's, Parkinson's, and Huntington's diseases that were long characterized by mechanistic ignorance have yielded striking progress in our understanding of their molecular underpinnings. A central theme in these and related disorders is the concept that certain normally soluble neuronal proteins can misfold and aggregate into oligomers and amyloid fibrils which can confer profound cytotoxicity. Perhaps the foremost example, both in terms of its societal impact and how far knowledge has moved toward the clinic, is that of Alzheimer's disease (AD). Here, we will review the classical protein lesions of the disorder that have provided a road map to etiology and pathogenesis. We will discuss how elucidating the genotype-to-phenotype relationships of familial forms of Alzheimer's disease has highlighted the importance of the misfolding and altered proteostasis of two otherwise soluble proteins, amyloid β-protein and tau, suggesting mechanism-based therapeutic targets that have led to clinical trials.
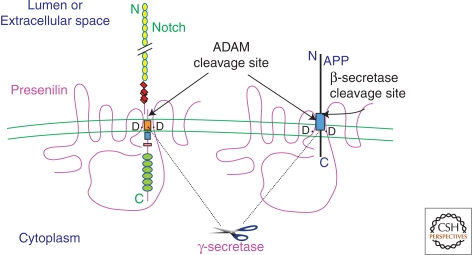
Figures


References
-
- Anliker B, Muller U 2006. The functions of mammalian amyloid precursor protein and related amyloid precursor-like proteins. Neurodegener Dis 3: 239–246 - PubMed
-
- Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, et al. 1997. Lack of apolipprotein E dramatically reduces amyloid β-peptide deposition. Nat Genet 17: 263–264 - PubMed
-
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, et al. 2000. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6: 916–919 - PubMed
-
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada C-M, Kim G, Seekins S, Yager D, et al. 1996. Familial Alzheimer’s disease–linked presenilin 1 variants elevate Aβ1-42/1-40 ratio in vitro and in vivo. Neuron 17: 1005–1013 - PubMed
-
- Brockhaus M, Grunberg J, Rohrig S, Loetscher H, Wittenburg N, Baumeister R, Jacobsen H, Haass C 1998. Caspasse-mediated cleavage is not required for the activity of presenilins in amyloidogenesis and NOTCH signaling. NeuroReport 9: 1481–1486 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical