

Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress
- PMID: 21576264
- PMCID: PMC3093120
- DOI: 10.1101/gad.1987211
Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress
Abstract
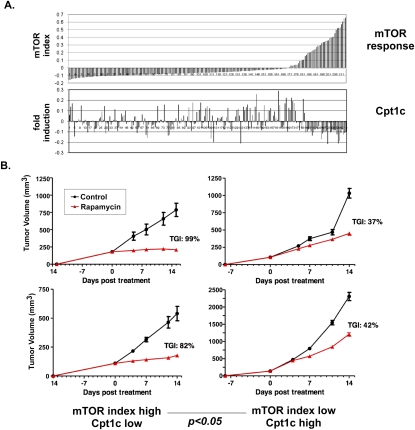
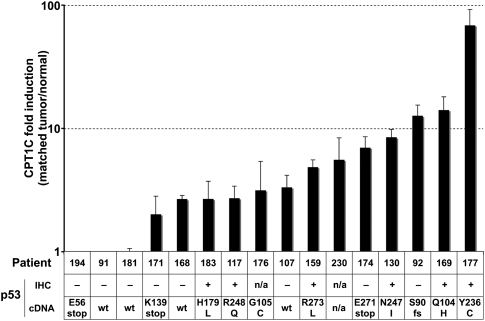
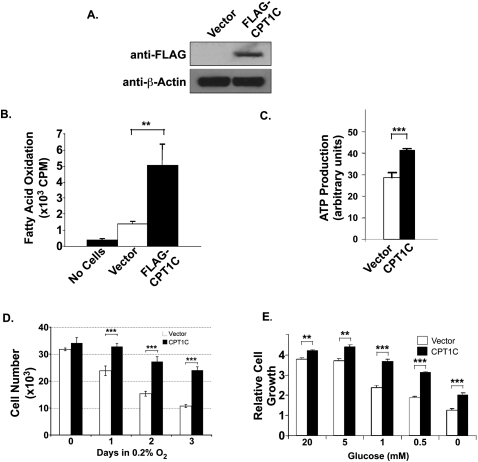
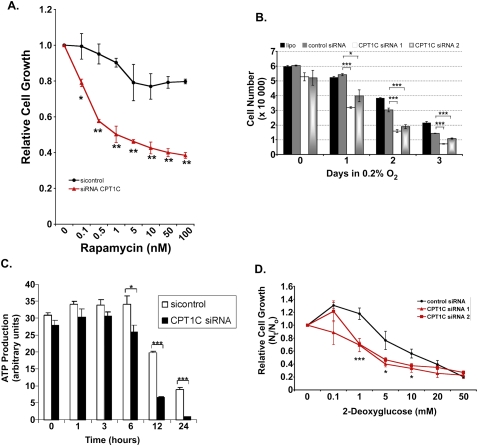
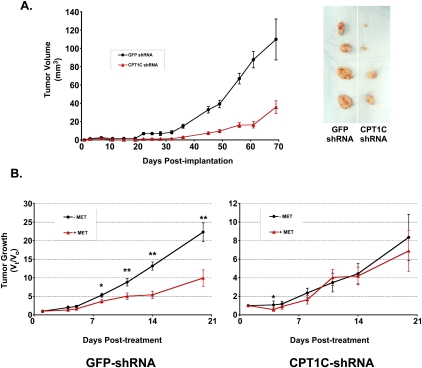
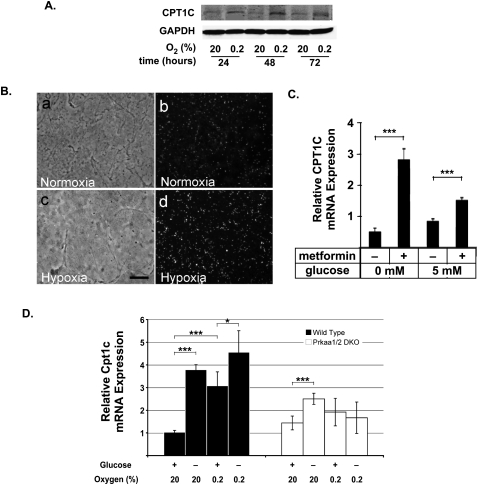
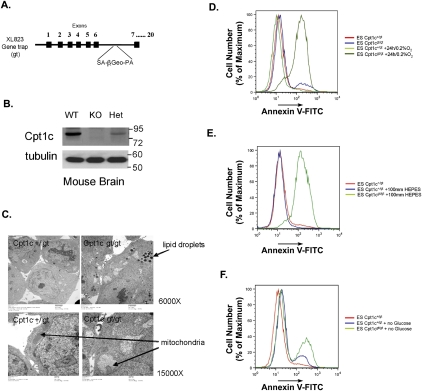
Tumor cells gain a survival/growth advantage by adapting their metabolism to respond to environmental stress, a process known as metabolic transformation. The best-known aspect of metabolic transformation is the Warburg effect, whereby cancer cells up-regulate glycolysis under aerobic conditions. However, other mechanisms mediating metabolic transformation remain undefined. Here we report that carnitine palmitoyltransferase 1C (CPT1C), a brain-specific metabolic enzyme, may participate in metabolic transformation. CPT1C expression correlates inversely with mammalian target of rapamycin (mTOR) pathway activation, contributes to rapamycin resistance in murine primary tumors, and is frequently up-regulated in human lung tumors. Tumor cells constitutively expressing CPT1C show increased fatty acid (FA) oxidation, ATP production, and resistance to glucose deprivation or hypoxia. Conversely, cancer cells lacking CPT1C produce less ATP and are more sensitive to metabolic stress. CPT1C depletion via siRNA suppresses xenograft tumor growth and metformin responsiveness in vivo. CPT1C can be induced by hypoxia or glucose deprivation and is regulated by AMPKα. Cpt1c-deficient murine embryonic stem (ES) cells show sensitivity to hypoxia and glucose deprivation and altered FA homeostasis. Our results indicate that cells can use a novel mechanism involving CPT1C and FA metabolism to protect against metabolic stress. CPT1C may thus be a new therapeutic target for the treatment of hypoxic tumors.
Figures
References
-
- Brown JM, Wilson WR 2004. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer 4: 437–447 - PubMed
-
- Buck E, Eyzaguirre A, Brown E, Petti F, McCormack S, Haley JD, Iwata KK, Gibson NW, Griffin G 2006. Rapamycin synergizes with the epidermal growth factor receptor inhibitor erlotinib in non-small-cell lung, pancreatic, colon, and breast tumors. Mol Cancer Ther 5: 2676–2684 - PubMed
-
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B 1998. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282: 1497–1501 - PubMed
-
- Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB 2007. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res 67: 6745–6752 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous