

Viral myocarditis: potential defense mechanisms within the cardiomyocyte against virus infection
- PMID: 21585262
- PMCID: PMC3131135
- DOI: 10.2217/fmb.11.40
Viral myocarditis: potential defense mechanisms within the cardiomyocyte against virus infection
Abstract
Virus infection can inflict significant damage on cardiomyocytes through direct injury and secondary immune reactions, leading to myocarditis and dilated cardiomyopathy. While viral myocarditis or cardiomyopathy is a complication of systemic infection of cardiotropic viruses, most individuals infected with the viruses do not develop significant cardiac disease. However, some individuals proceed to develop severe virus-mediated heart disease. Recent studies have shown that viral infection of cardiomyocytes is required for the development of myocarditis and subsequent cardiomyopathy. This suggests that viral infection of cardiomyocytes can be an important step that determines the pathogenesis of viral myocarditis during systemic infection. Accordingly, this article focuses on potential defense mechanisms within the cardiomyocyte against virus infection. Understanding of the cardiomyocyte defense against invading viruses may give us novel insights into the pathophysiology of viral myocarditis, and enable us to develop innovative strategies of diagnosis and treatment for this challenging clinical entity.
Figures
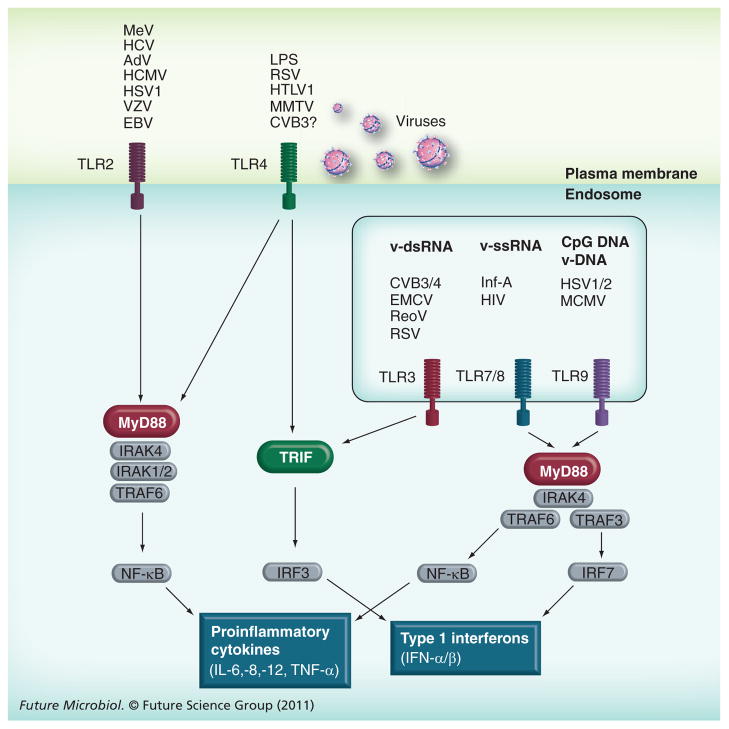
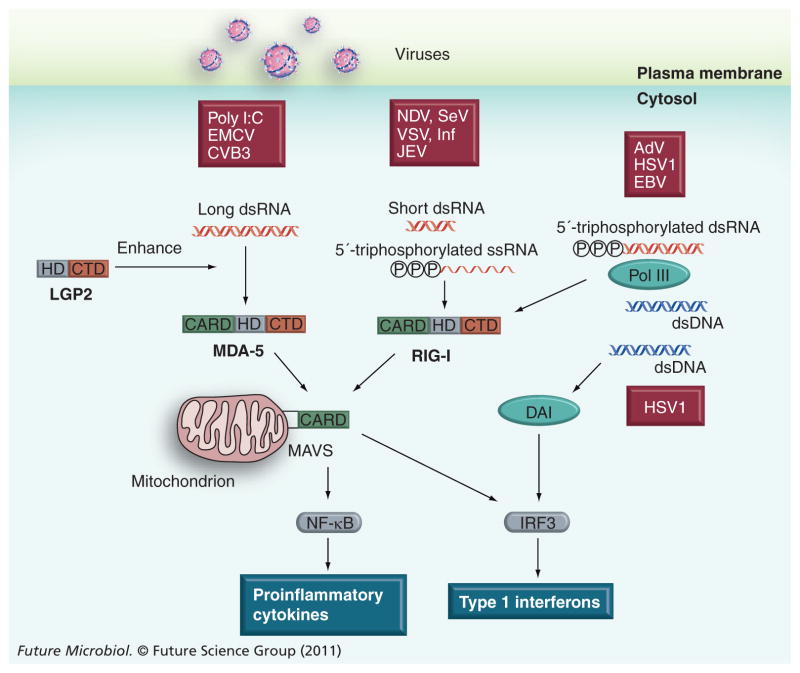
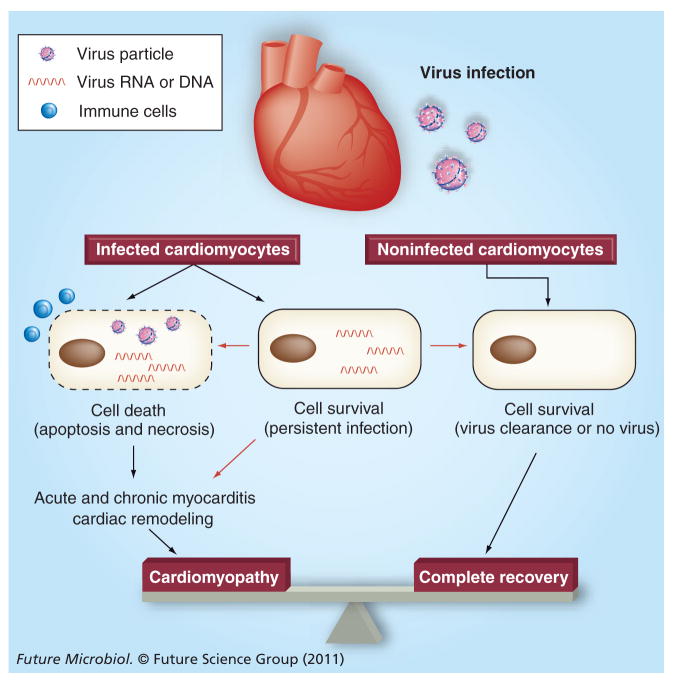
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical