

A fast and accurate method to detect allelic genomic imbalances underlying mosaic rearrangements using SNP array data
- PMID: 21586113
- PMCID: PMC3118168
- DOI: 10.1186/1471-2105-12-166
A fast and accurate method to detect allelic genomic imbalances underlying mosaic rearrangements using SNP array data
Abstract
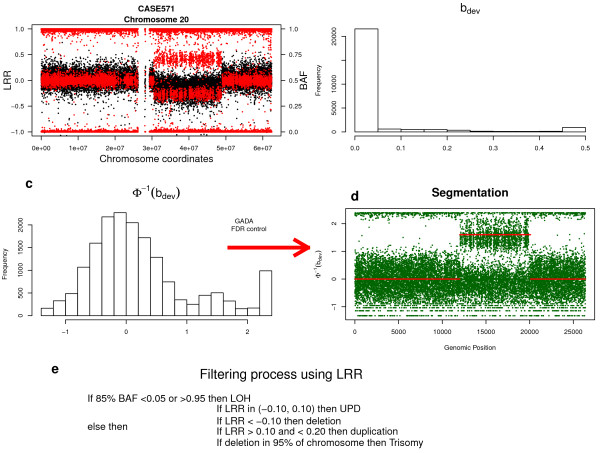
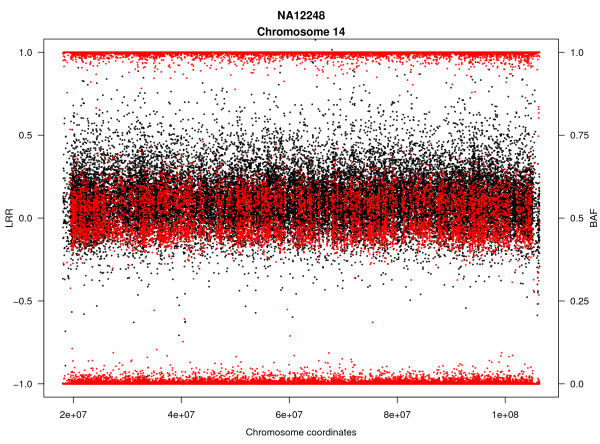
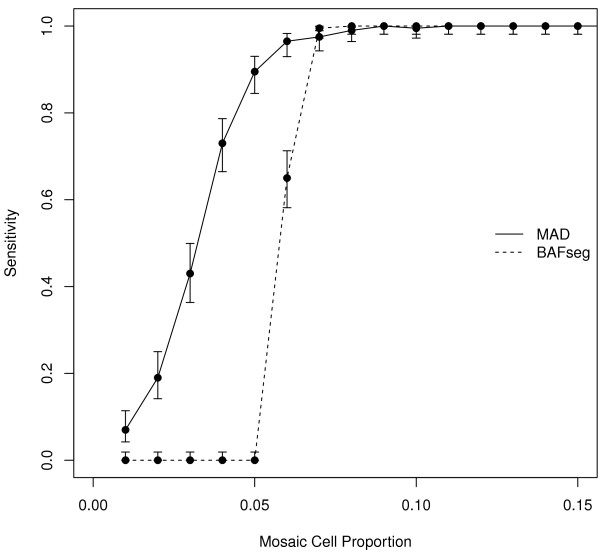
Background: Mosaicism for copy number and copy neutral chromosomal rearrangements has been recently identified as a relatively common source of genetic variation in the normal population. However its prevalence is poorly defined since it has been only studied systematically in one large-scale study and by using non optimal ad-hoc SNP array data analysis tools, uncovering rather large alterations (> 1 Mb) and affecting a high proportion of cells. Here we propose a novel methodology, Mosaic Alteration Detection-MAD, by providing a software tool that is effective for capturing previously described alterations as wells as new variants that are smaller in size and/or affecting a low percentage of cells.
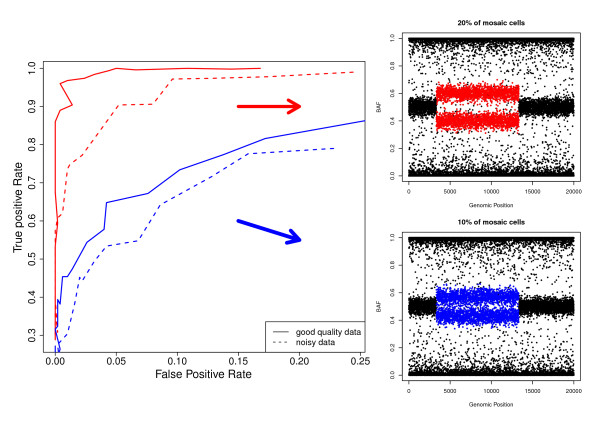
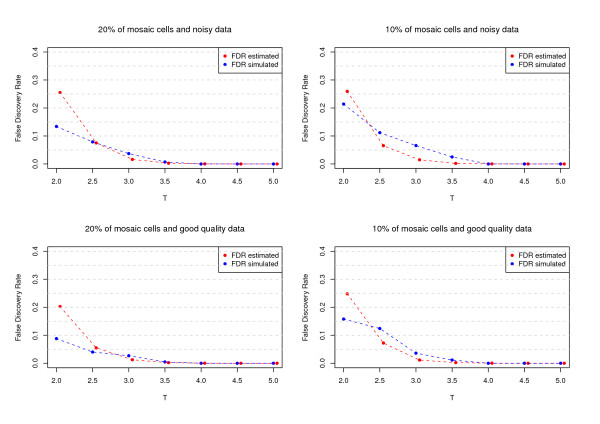
Results: The developed method identified all previously known mosaic abnormalities reported in SNP array data obtained from controls, bladder cancer and HapMap individuals. In addition MAD tool was able to detect new mosaic variants not reported before that were smaller in size and with lower percentage of cells affected. The performance of the tool was analysed by studying simulated data for different scenarios. Our method showed high sensitivity and specificity for all assessed scenarios.
Conclusions: The tool presented here has the ability to identify mosaic abnormalities with high sensitivity and specificity. Our results confirm the lack of sensitivity of former methods by identifying new mosaic variants not reported in previously utilised datasets. Our work suggests that the prevalence of mosaic alterations could be higher than initially thought. The use of appropriate SNP array data analysis methods would help in defining the human genome mosaic map.
Figures
References
-
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M. Large-scale copy number polymorphism in the human genome. Science. 2004;305(5683):525–8. doi: 10.1126/science.1098918. - DOI - PubMed
-
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–54. doi: 10.1038/nature05329. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
