

Genome-wide evidence for local DNA methylation spreading from small RNA-targeted sequences in Arabidopsis
- PMID: 21586580
- PMCID: PMC3167636
- DOI: 10.1093/nar/gkr324
Genome-wide evidence for local DNA methylation spreading from small RNA-targeted sequences in Arabidopsis
Abstract
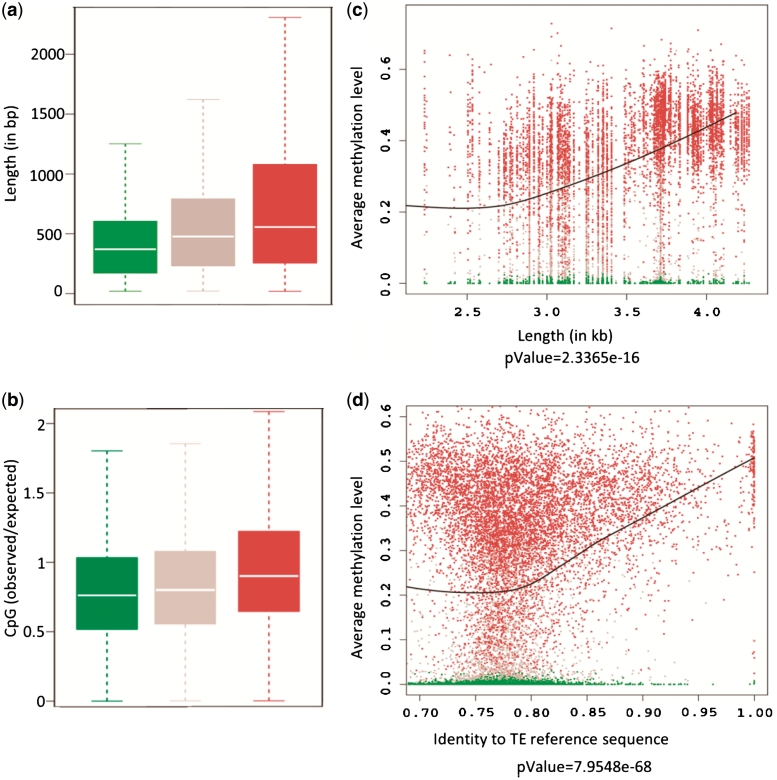
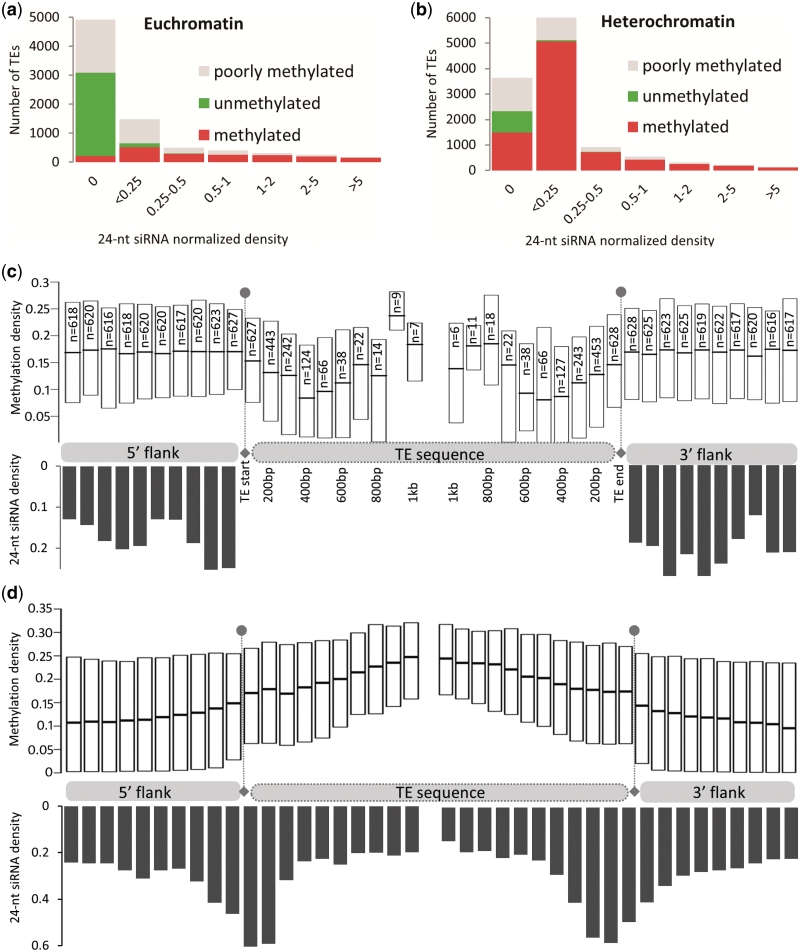
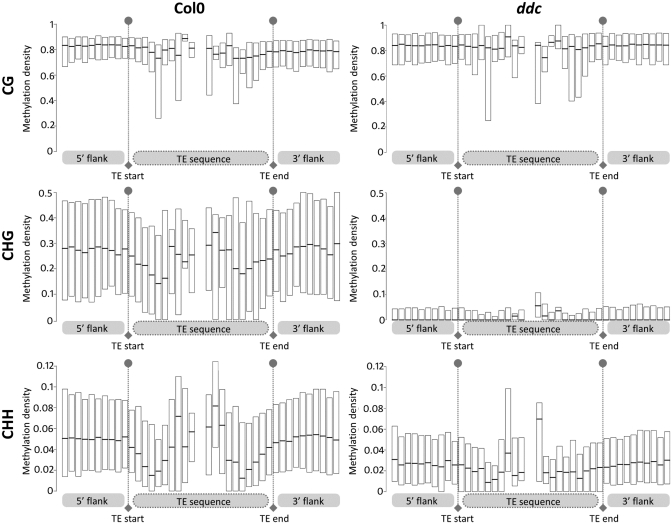
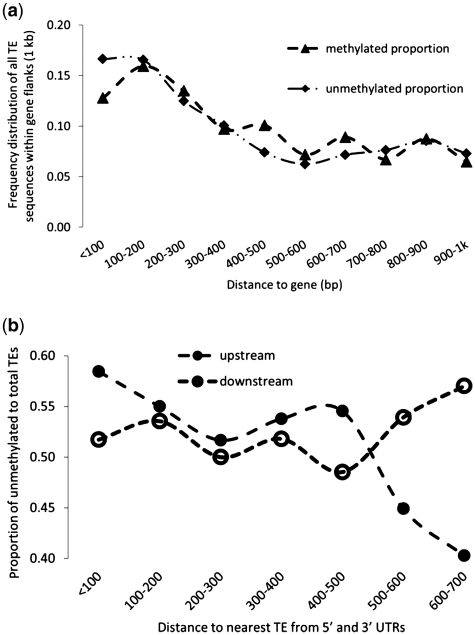
Transposable elements (TEs) and their relics play major roles in genome evolution. However, mobilization of TEs is usually deleterious and strongly repressed. In plants and mammals, this repression is typically associated with DNA methylation, but the relationship between this epigenetic mark and TE sequences has not been investigated systematically. Here, we present an improved annotation of TE sequences and use it to analyze genome-wide DNA methylation maps obtained at single-nucleotide resolution in Arabidopsis. We show that although the majority of TE sequences are methylated, ∼26% are not. Moreover, a significant fraction of TE sequences densely methylated at CG, CHG and CHH sites (where H = A, T or C) have no or few matching small interfering RNA (siRNAs) and are therefore unlikely to be targeted by the RNA-directed DNA methylation (RdDM) machinery. We provide evidence that these TE sequences acquire DNA methylation through spreading from adjacent siRNA-targeted regions. Further, we show that although both methylated and unmethylated TE sequences located in euchromatin tend to be more abundant closer to genes, this trend is least pronounced for methylated, siRNA-targeted TE sequences located 5' to genes. Based on these and other findings, we propose that spreading of DNA methylation through promoter regions explains at least in part the negative impact of siRNA-targeted TE sequences on neighboring gene expression.
Figures
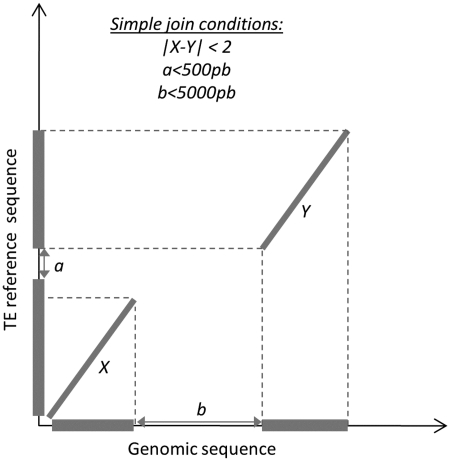
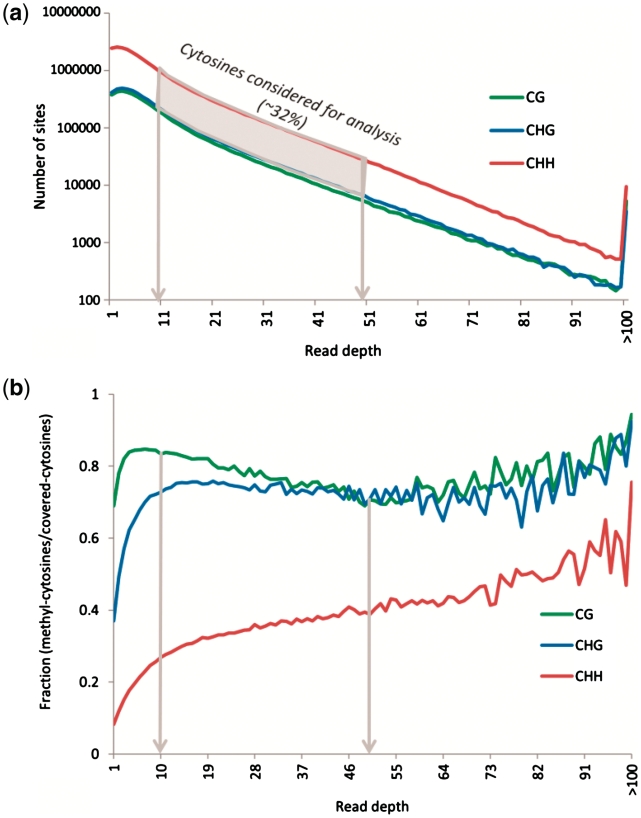
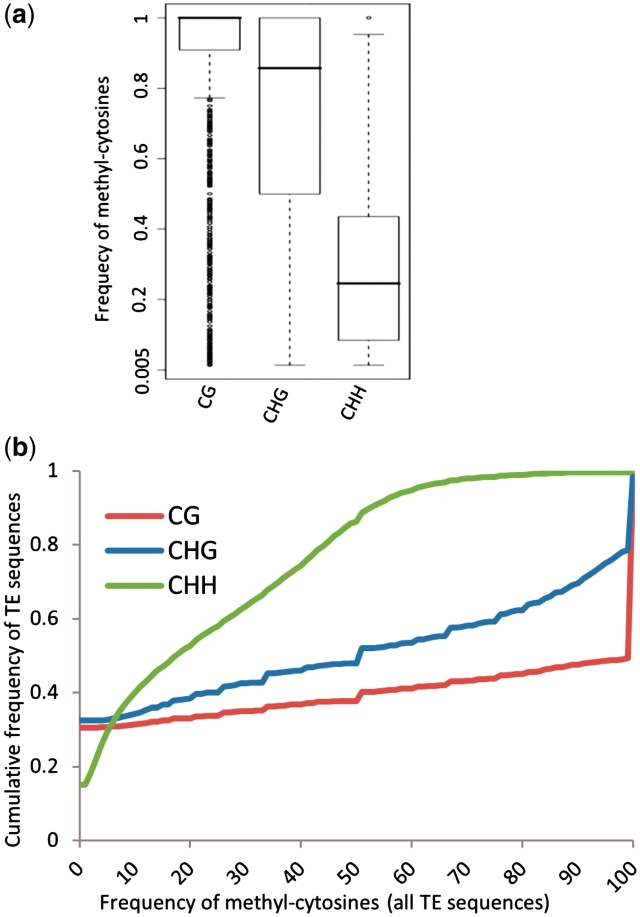
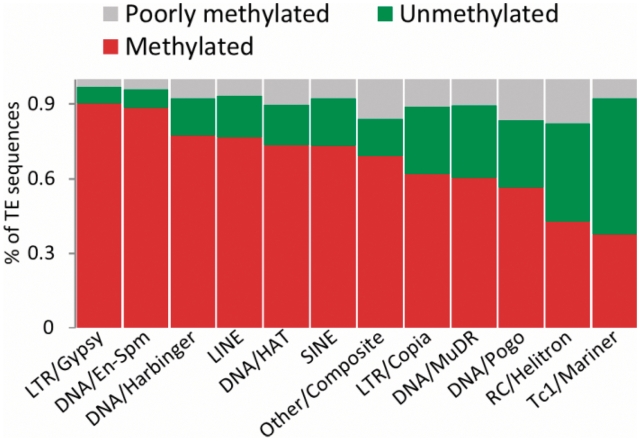
References
-
- Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007;8:272–285. - PubMed
-
- Teixeira FK, Colot V. Repeat elements and the Arabidopsis DNA methylation landscape. Heredity. 2010;105:14–23. - PubMed
-
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
