

Predicting sites of ADAR editing in double-stranded RNA
- PMID: 21587236
- PMCID: PMC3113232
- DOI: 10.1038/ncomms1324
Predicting sites of ADAR editing in double-stranded RNA
Abstract
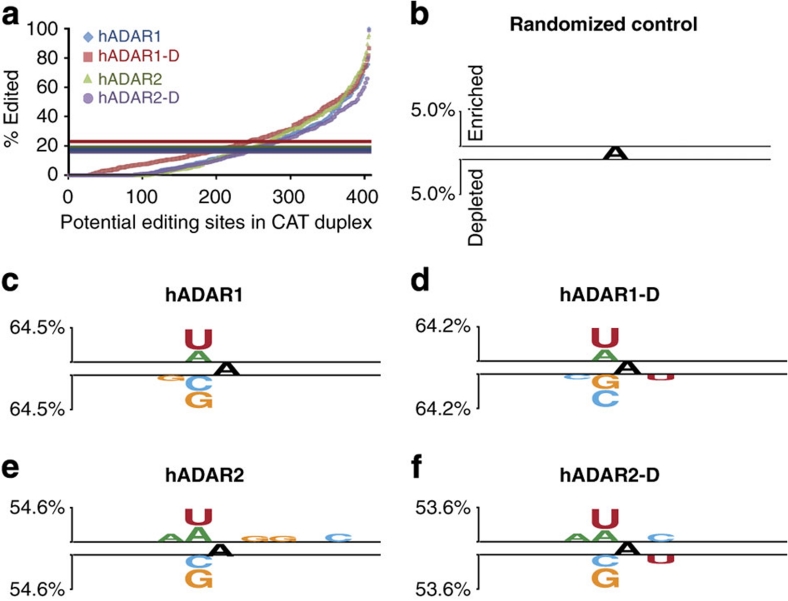
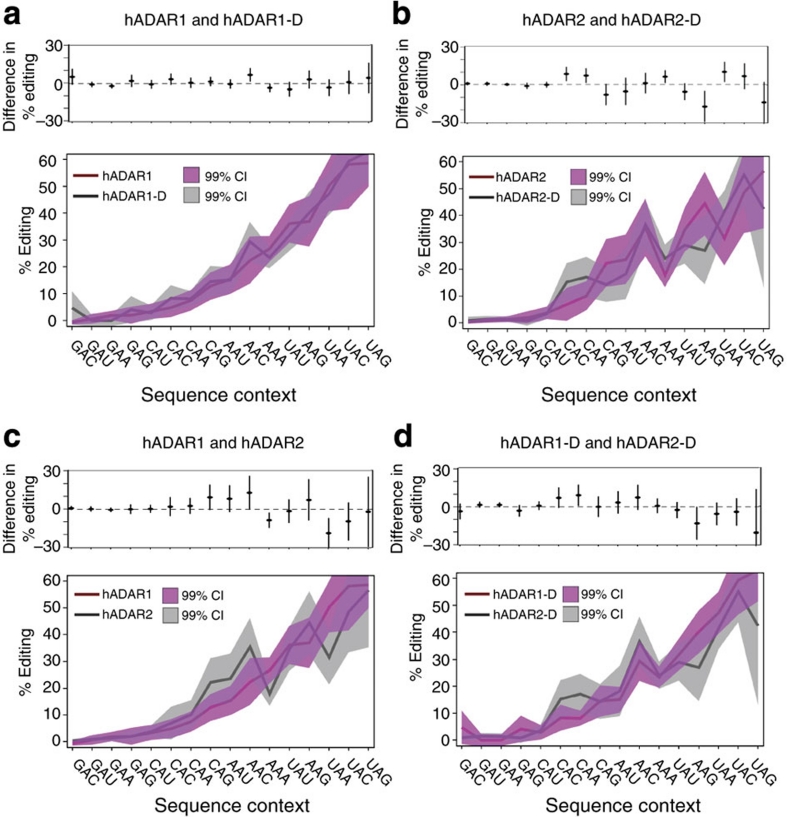
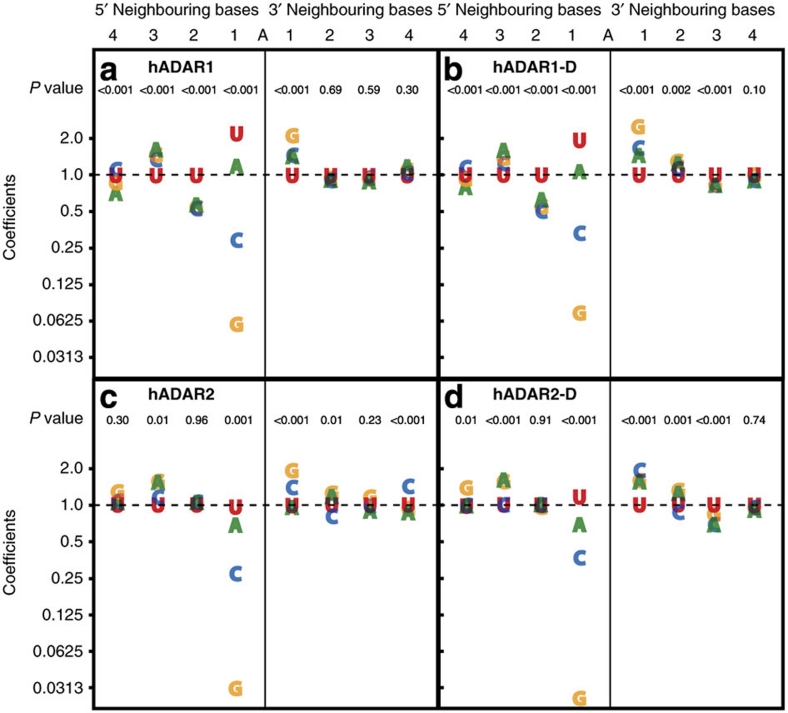
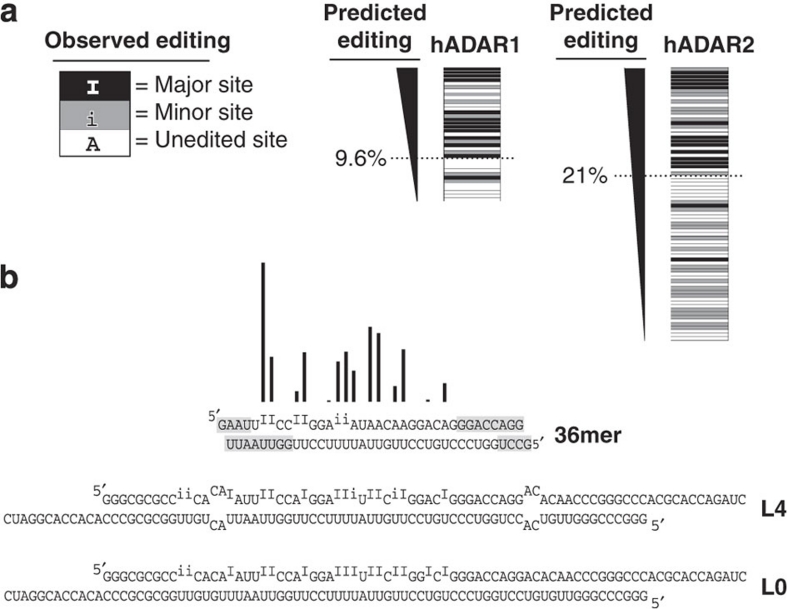
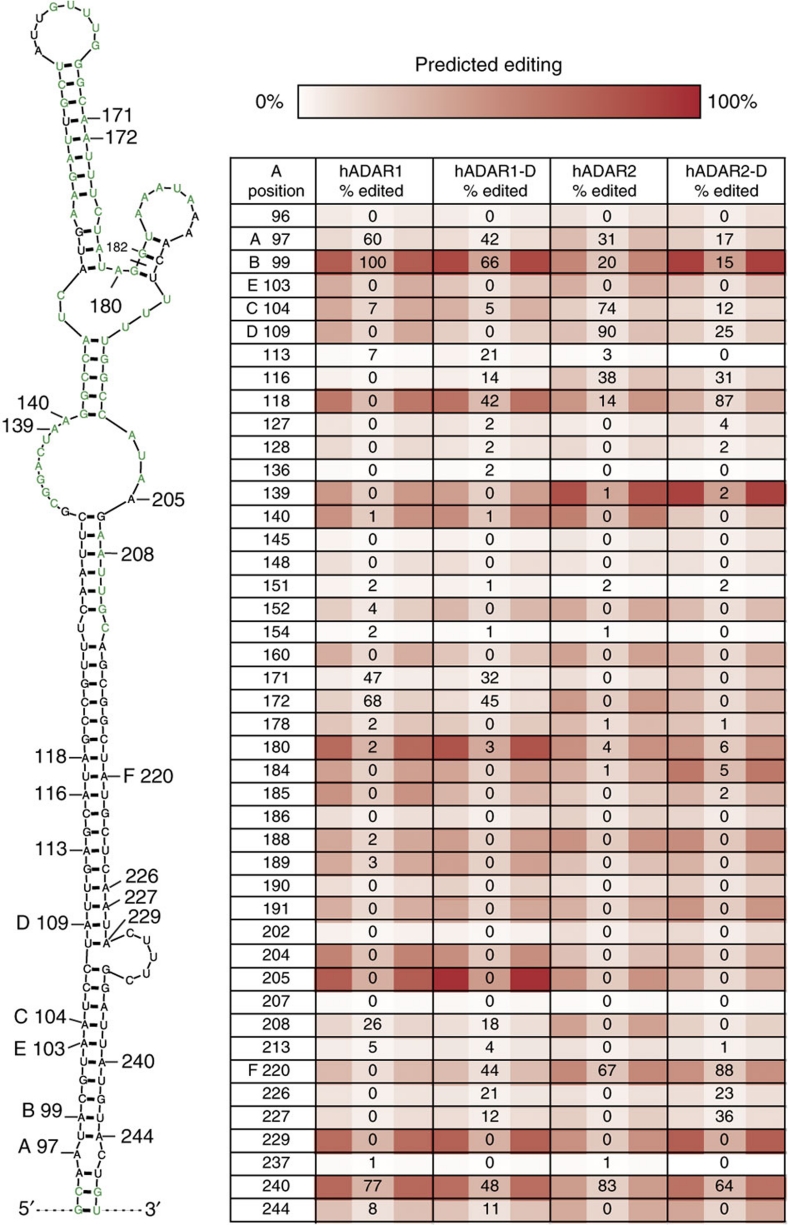
ADAR (adenosine deaminase that acts on RNA) editing enzymes target coding and noncoding double-stranded RNA (dsRNA) and are essential for neuronal function. Early studies showed that ADARs preferentially target adenosines with certain 5' and 3' neighbours. Here we use current Sanger sequencing protocols to perform a more accurate and quantitative analysis. We quantified editing sites in an ∼800-bp dsRNA after reaction with human ADAR1 or ADAR2, or their catalytic domains alone. These large data sets revealed that neighbour preferences are mostly dictated by the catalytic domain, but ADAR2's dsRNA-binding motifs contribute to 3' neighbour preferences. For all proteins, the 5' nearest neighbour was most influential, but adjacent bases also affected editing site choice. We developed algorithms to predict editing sites in dsRNA of any sequence, and provide a web-based application. The predictive power of the algorithm on fully base-paired dsRNA, compared with biological substrates containing mismatches, bulges and loops, elucidates structural contributions to editing specificity.
Figures
References
-
- Levanon E. Y. et al.. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 22 1001–1005 (2004). - PubMed
-
- Li J. B. et al.. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 324 1210–1213 (2009). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
