

Targeted assembly of short sequence reads
- PMID: 21589938
- PMCID: PMC3092772
- DOI: 10.1371/journal.pone.0019816
Targeted assembly of short sequence reads
Abstract
As next-generation sequence (NGS) production continues to increase, analysis is becoming a significant bottleneck. However, in situations where information is required only for specific sequence variants, it is not necessary to assemble or align whole genome data sets in their entirety. Rather, NGS data sets can be mined for the presence of sequence variants of interest by localized assembly, which is a faster, easier, and more accurate approach. We present TASR, a streamlined assembler that interrogates very large NGS data sets for the presence of specific variants by only considering reads within the sequence space of input target sequences provided by the user. The NGS data set is searched for reads with an exact match to all possible short words within the target sequence, and these reads are then assembled stringently to generate a consensus of the target and flanking sequence. Typically, variants of a particular locus are provided as different target sequences, and the presence of the variant in the data set being interrogated is revealed by a successful assembly outcome. However, TASR can also be used to find unknown sequences that flank a given target. We demonstrate that TASR has utility in finding or confirming genomic mutations, polymorphisms, fusions and integration events. Targeted assembly is a powerful method for interrogating large data sets for the presence of sequence variants of interest. TASR is a fast, flexible and easy to use tool for targeted assembly.
Conflict of interest statement
Figures
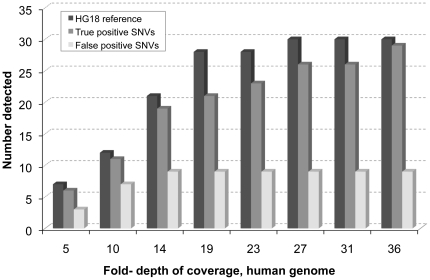
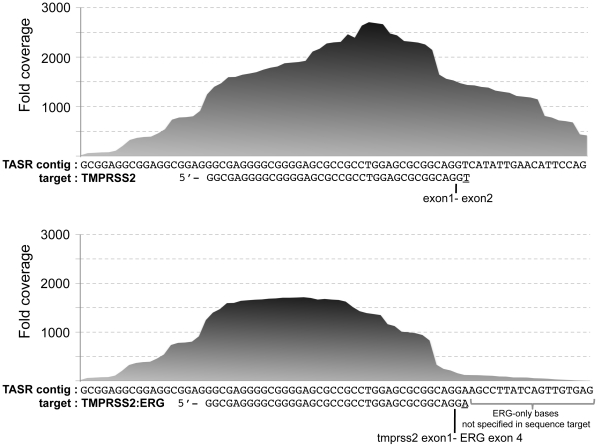
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
