

Long-term safety and efficacy of deferasirox (Exjade) for up to 5 years in transfusional iron-overloaded patients with sickle cell disease
- PMID: 21592110
- PMCID: PMC3170481
- DOI: 10.1111/j.1365-2141.2011.08720.x
Long-term safety and efficacy of deferasirox (Exjade) for up to 5 years in transfusional iron-overloaded patients with sickle cell disease
Abstract
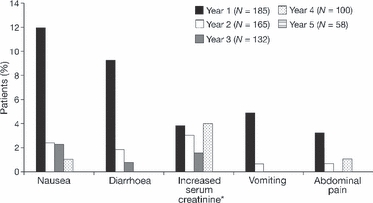
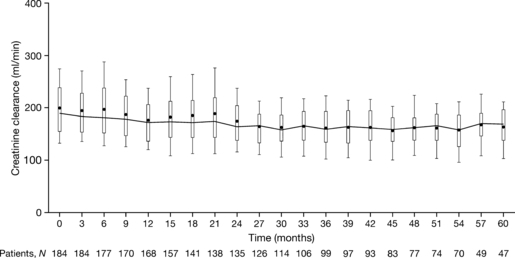
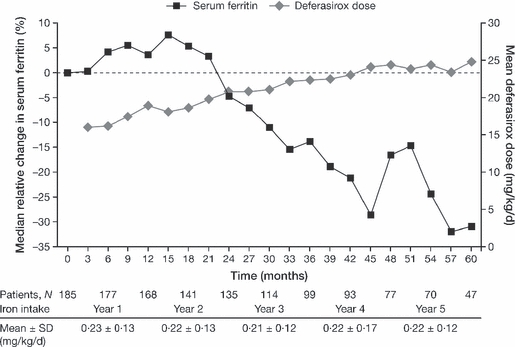
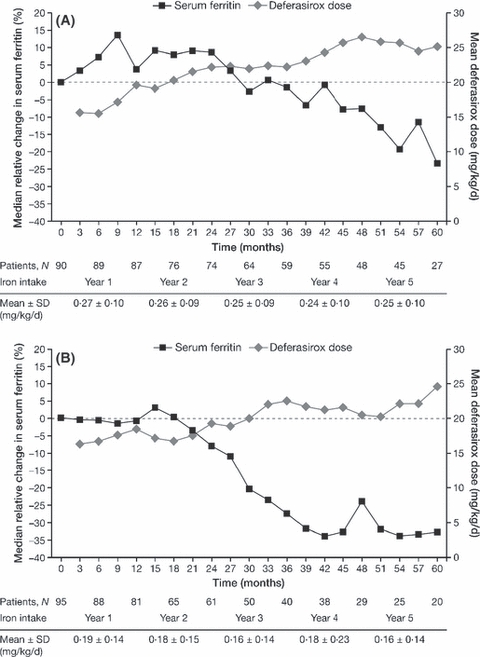
To date, there is a lack of long-term safety and efficacy data for iron chelation therapy in transfusion-dependent patients with sickle cell disease (SCD). To evaluate the long-term safety and efficacy of deferasirox (a once-daily oral iron chelator), patients with SCD completing a 1-year, Phase II, randomized, deferoxamine (DFO)-controlled study entered a 4-year extension, continuing to receive deferasirox, or switching from DFO to deferasirox. Average actual deferasirox dose was 19·4 ± 6·3 mg/kg per d. Of 185 patients who received at least one deferasirox dose, 33·5% completed the 5-year study. The most common reasons for discontinuation were withdrawal of consent (23·8%), lost to follow-up (9·2%) and adverse events (AEs) (7·6%). Investigator-assessed drug-related AEs were predominantly gastrointestinal [including nausea (14·6%), diarrhoea (10·8%)], mild-to-moderate and transient in nature. Creatinine clearance remained within the normal range throughout the study. Despite conservative initial dosing, serum ferritin levels in patients with ≥ 4 years deferasirox exposure significantly decreased by -591 μg/l (95% confidence intervals, -1411, -280 μg/l; P = 0·027; n = 67). Long-term deferasirox treatment for up to 5 years had a clinically acceptable safety profile, including maintenance of normal renal function, in patients with SCD. Iron burden was substantially reduced with appropriate dosing in patients treated for at least 4 years.
© 2011 Blackwell Publishing Ltd.
Figures
Comment in
-
Safety of deferasirox in sickle cell disease patients with co-existing liver impairment.Br J Haematol. 2012 May;157(4):505-6; author reply 506-7. doi: 10.1111/j.1365-2141.2012.09040.x. Epub 2012 Feb 2. Br J Haematol. 2012. PMID: 22299817 No abstract available.
References
-
- Adams RJ, Brambilla D. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. New England Journal of Medicine. 2005;353:2769–2778. - PubMed
-
- Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, Abboud M, Gallagher D, Kutlar A, Nichols FT, Bonds DR, Brambilla D. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. New England Journal of Medicine. 1998;339:5–11. - PubMed
-
- Ballas SK. Iron overload is a determinant of morbidity and mortality in adult patients with sickle cell disease. Seminars in Hematology. 2001;38(Suppl. 1):30–36. - PubMed
-
- Brittenham GM, Cohen AR, McLaren CE, Martin MB, Griffith PM, Nienhuis AW, Young NS, Allen CJ, Farrell DE, Harris JW. Hepatic iron stores and plasma ferritin concentration in patients with sickle cell anemia and thalassemia major. American Journal of Hematology. 1993;42:81–85. - PubMed
-
- Brown K, Subramony C, May W, Megason G, Liu H, Bishop P, Walker T, Nowicki MJ. Hepatic iron overload in children with sickle cell anemia on chronic transfusion therapy. Journal of Pediatric Hematology/oncology. 2009;31:309–312. - PubMed
