

Demographic processes affect HIV-1 evolution in primary infection before the onset of selective processes
- PMID: 21593162
- PMCID: PMC3147913
- DOI: 10.1128/JVI.02697-10
Demographic processes affect HIV-1 evolution in primary infection before the onset of selective processes
Abstract
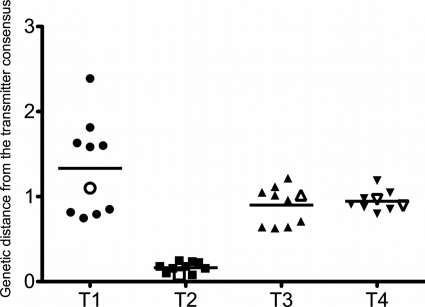
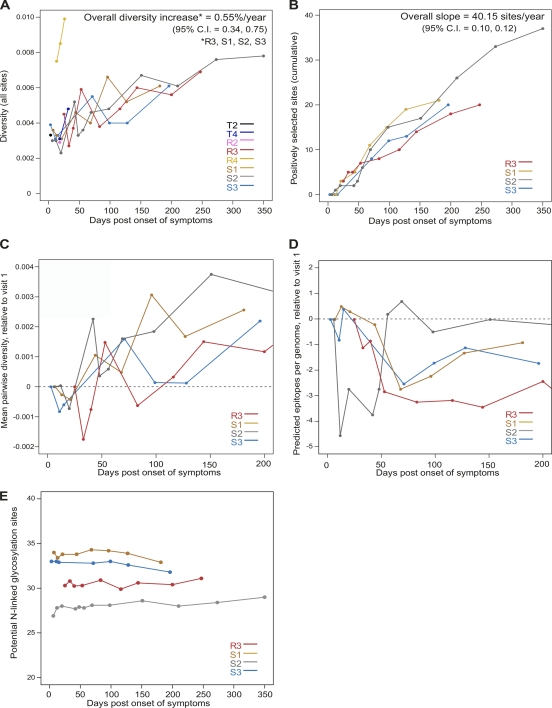
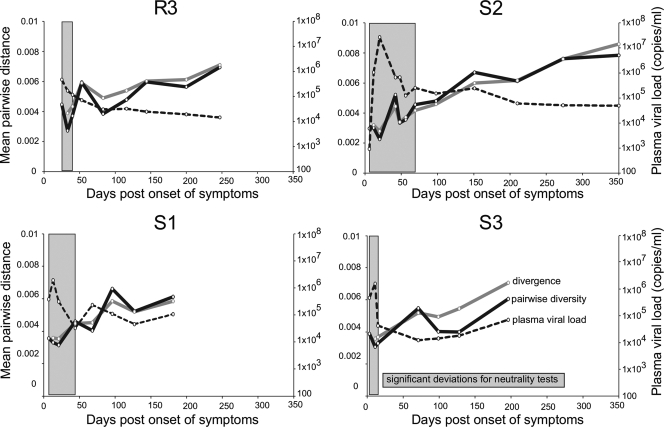
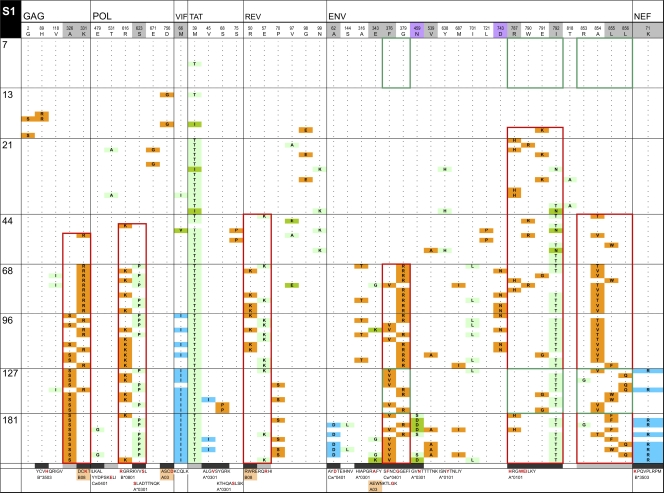
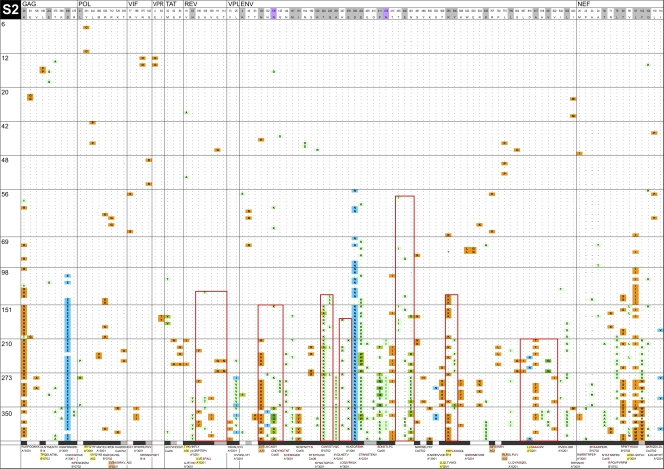
HIV-1 transmission and viral evolution in the first year of infection were studied in 11 individuals representing four transmitter-recipient pairs and three independent seroconverters. Nine of these individuals were enrolled during acute infection; all were men who have sex with men (MSM) infected with HIV-1 subtype B. A total of 475 nearly full-length HIV-1 genome sequences were generated, representing on average 10 genomes per specimen at 2 to 12 visits over the first year of infection. Single founding variants with nearly homogeneous viral populations were detected in eight of the nine individuals who were enrolled during acute HIV-1 infection. Restriction to a single founder variant was not due to a lack of diversity in the transmitter as homogeneous populations were found in recipients from transmitters with chronic infection. Mutational patterns indicative of rapid viral population growth dominated during the first 5 weeks of infection and included a slight contraction of viral genetic diversity over the first 20 to 40 days. Subsequently, selection dominated, most markedly in env and nef. Mutants were detected in the first week and became consensus as early as day 21 after the onset of symptoms of primary HIV infection. We found multiple indications of cytotoxic T lymphocyte (CTL) escape mutations while reversions appeared limited. Putative escape mutations were often rapidly replaced with mutually exclusive mutations nearby, indicating the existence of a maturational escape process, possibly in adaptation to viral fitness constraints or to immune responses against new variants. We showed that establishment of HIV-1 infection is likely due to a biological mechanism that restricts transmission rather than to early adaptive evolution during acute infection. Furthermore, the diversity of HIV strains coupled with complex and individual-specific patterns of CTL escape did not reveal shared sequence characteristics of acute infection that could be harnessed for vaccine design.
Figures

References
-
- Bhattacharya T., et al. 2007. Founder effects in the assessment of HIV polymorphisms and HLA allele associations. Science 315:1583–1586 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
