

Abnormal trafficking and degradation of TLR4 underlie the elevated inflammatory response in cystic fibrosis
- PMID: 21593379
- PMCID: PMC3111054
- DOI: 10.4049/jimmunol.1100396
Abnormal trafficking and degradation of TLR4 underlie the elevated inflammatory response in cystic fibrosis
Abstract
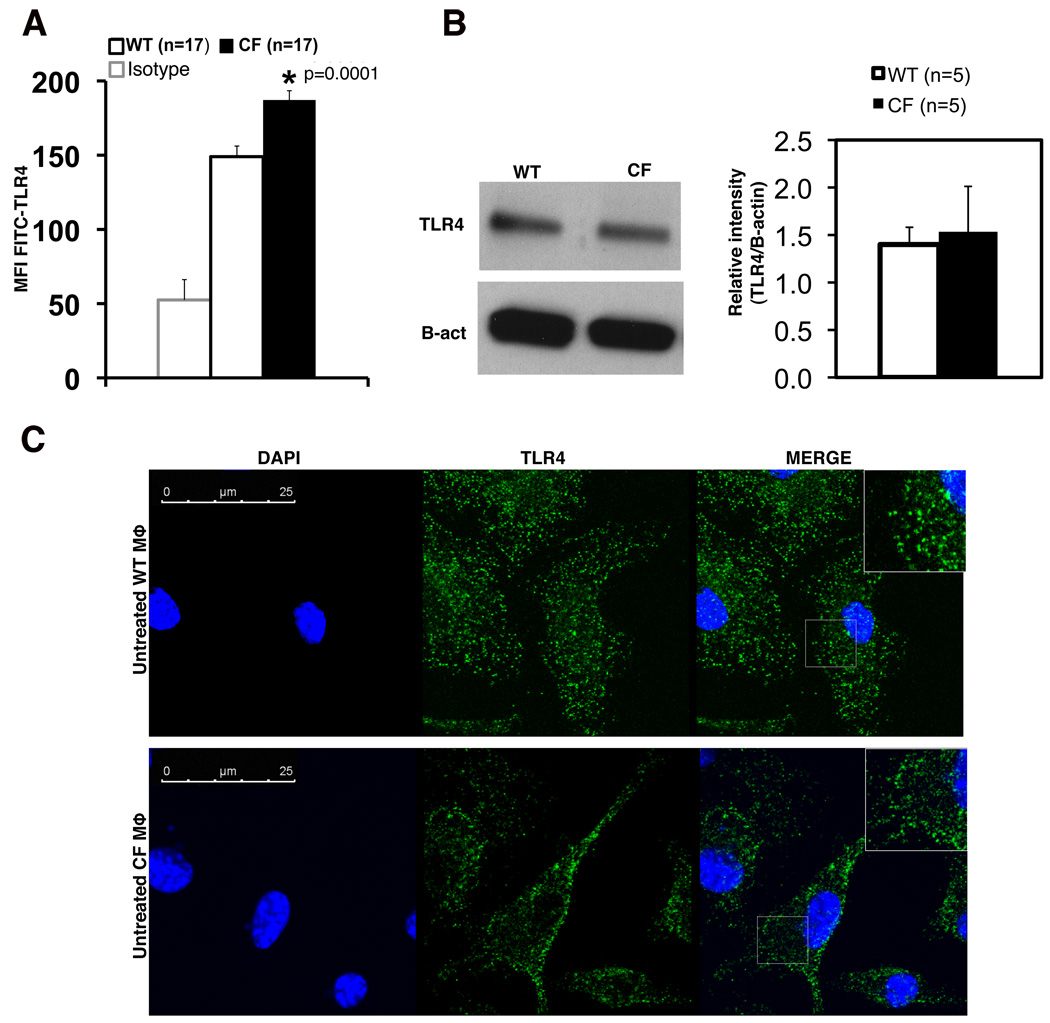
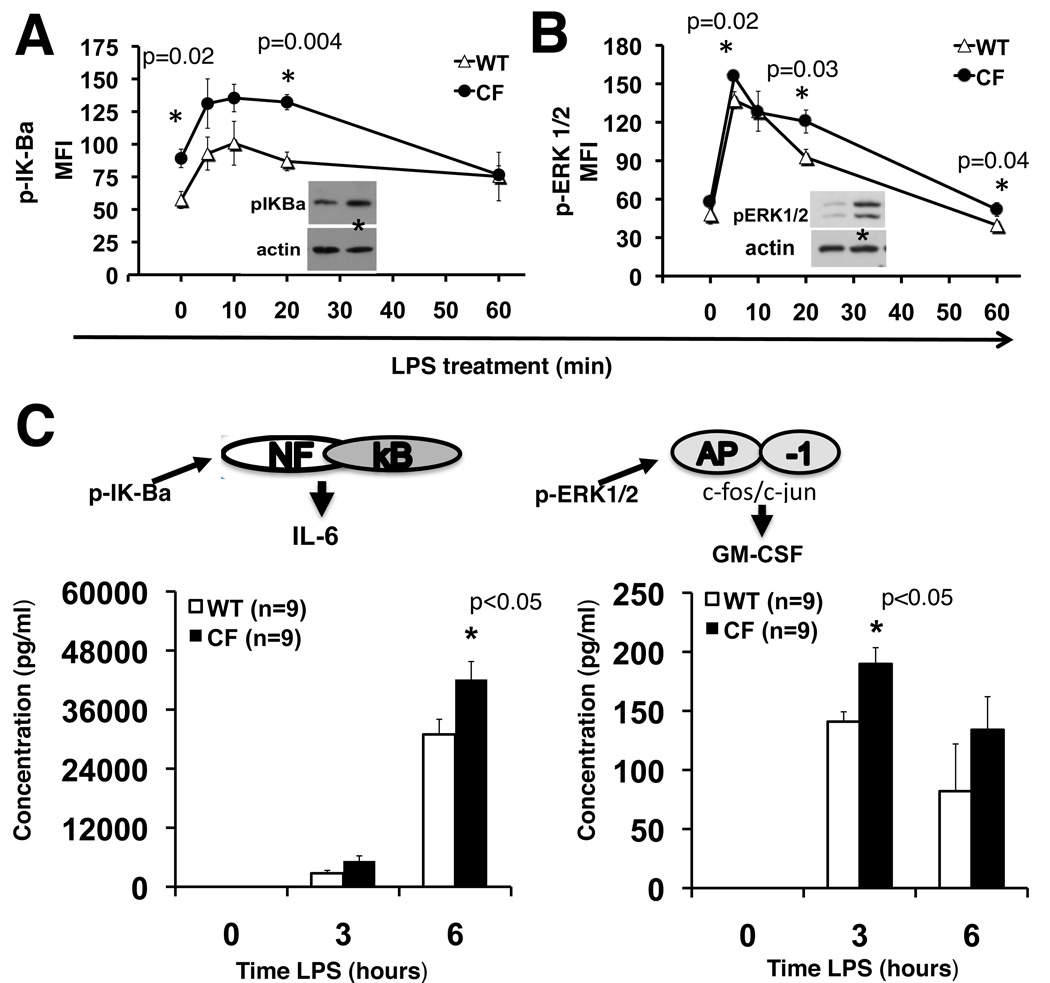
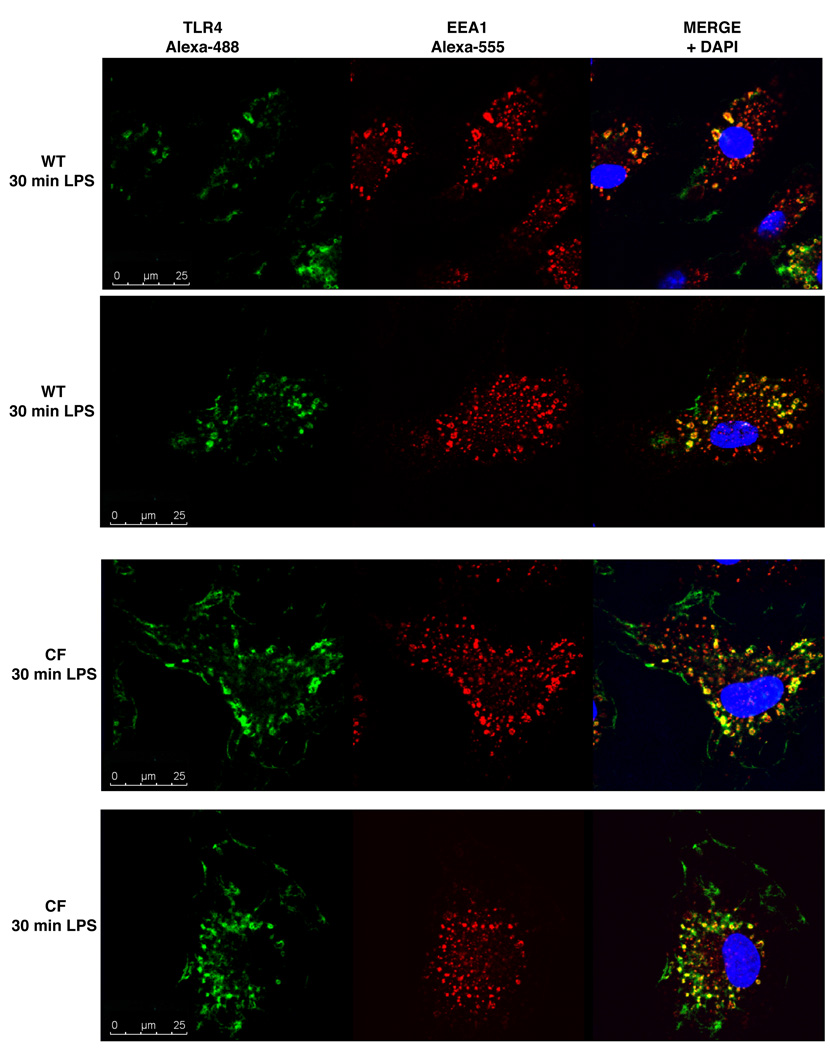
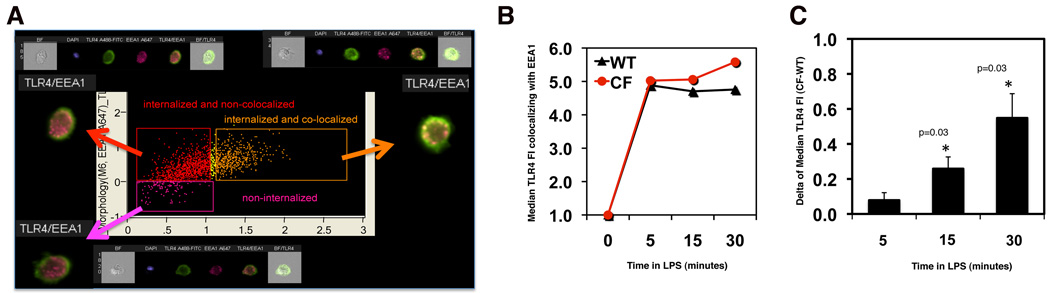
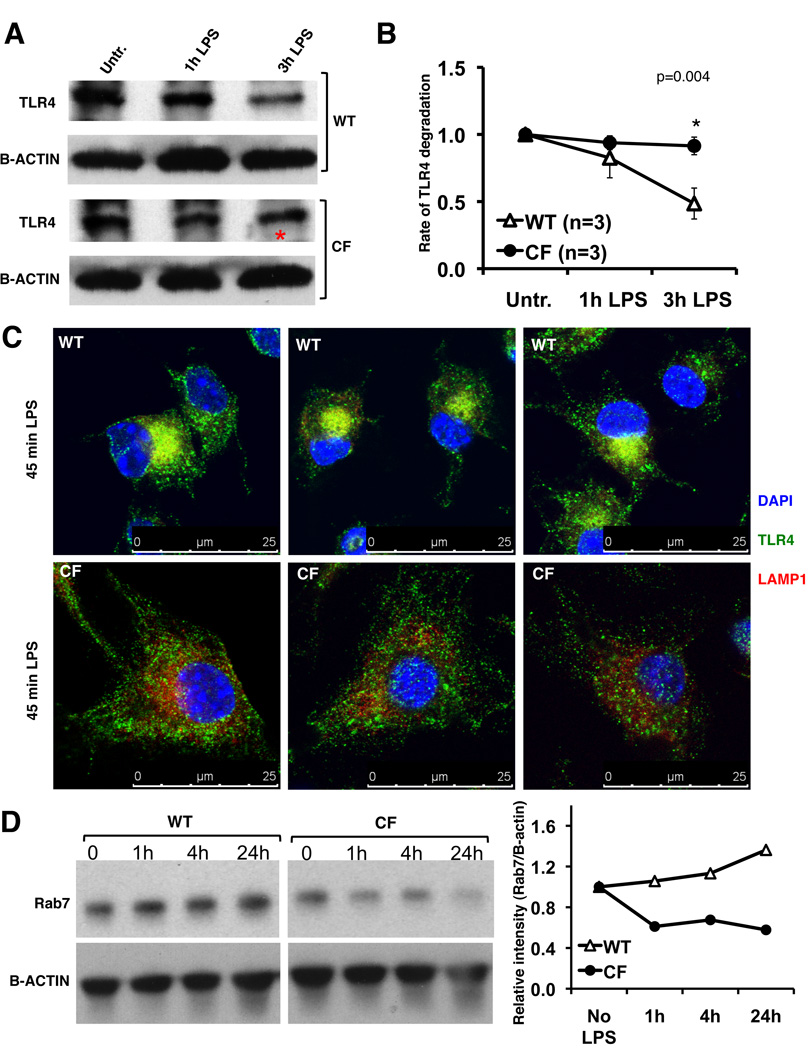
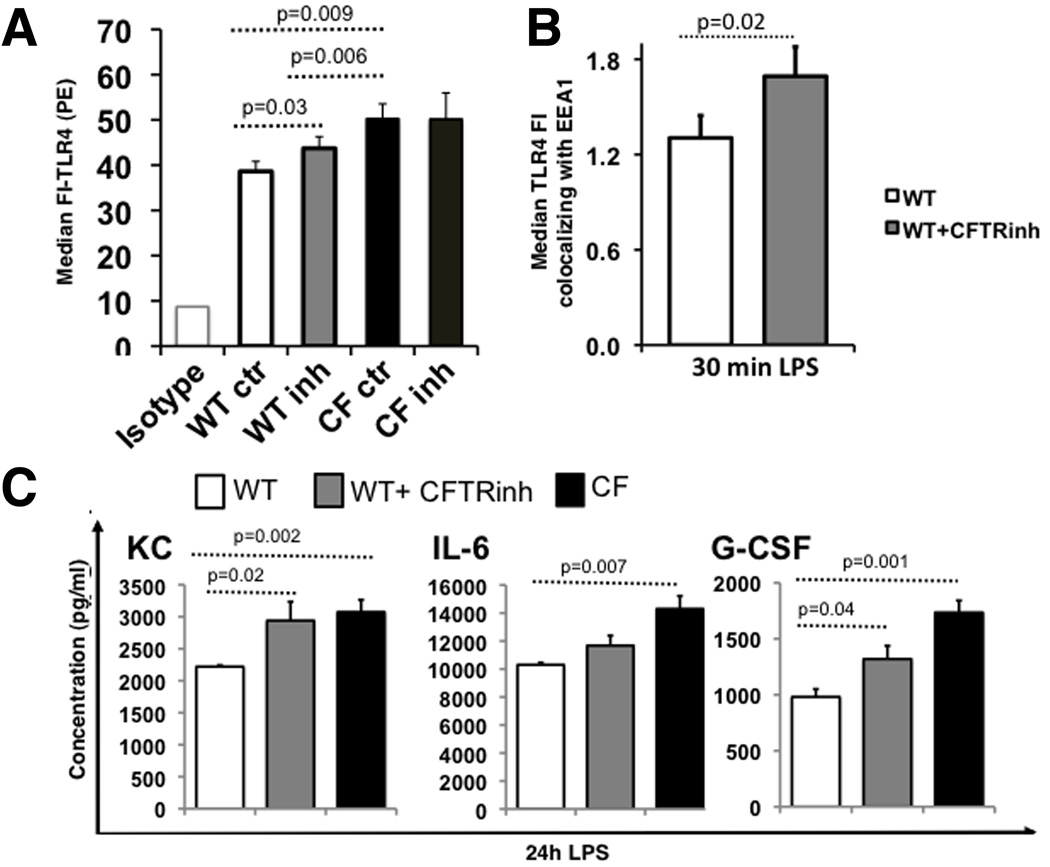
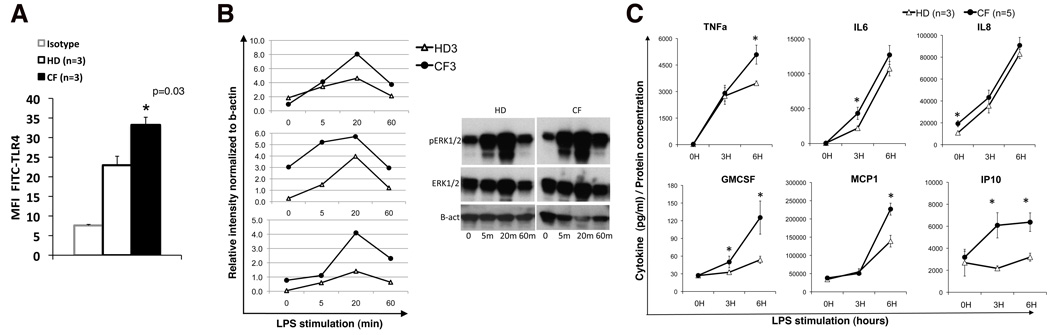
Morbidity and mortality in cystic fibrosis (CF) are due not only to abnormal epithelial cell function, but also to an abnormal immune response. We have shown previously that macrophages lacking CF transmembrane conductance regulator (CFTR), the gene mutated in CF, contribute significantly to the hyperinflammatory response observed in CF. In this study, we show that lack of functional CFTR in murine macrophages causes abnormal TLR4 subcellular localization. Upon LPS stimulation, CFTR macrophages have prolonged TLR4 retention in the early endosome and reduced translocation into the lysosomal compartment. This abnormal TLR4 trafficking leads to increased LPS-induced activation of the NF-κB, MAPK, and IFN regulatory factor-3 pathways and decreased TLR4 degradation, which affects downregulation of the proinflammatory state. In addition to primary murine cells, mononuclear cells isolated from CF patients demonstrate similar defects in response to LPS. Moreover, specific inhibition of CFTR function induces abnormal TLR4 trafficking and enhances the inflammatory response of wild-type murine cells to LPS. Thus, functional CFTR in macrophages influences TLR4 spatial and temporal localization and perturbs LPS-mediated signaling in both murine CF models and patients with CF.
Figures
References
-
- Hubeau C, Puchelle E, Gaillard D. Distinct pattern of immune cell population in the lung of human fetuses with cystic fibrosis. J Allergy Clin Immunol. 2001;108:524–529. - PubMed
-
- Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med. 1995;151:1075–1082. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
