

SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism
- PMID: 21596231
- PMCID: PMC9689753
- DOI: 10.1016/j.jacc.2010.09.084
SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism
Abstract
Objectives: The aim of this study was to discern the role of the cardiac voltage-gated sodium ion channel SCN5A in the etiology of dilated cardiomyopathy (DCM).
Background: Dilated cardiomyopathy associates with mutations in the SCN5A gene, but the frequency, phenotype, and causative nature of these associations remain the focus of ongoing investigation.
Methods: Since 1991, DCM probands and family members have been enrolled in the Familial Cardiomyopathy Registry and extensively evaluated by clinical phenotype. Genomic deoxyribonucleic acid samples from 338 individuals among 289 DCM families were obtained and screened for SCN5A mutations by denaturing high-performance liquid chromatography and sequence analysis.
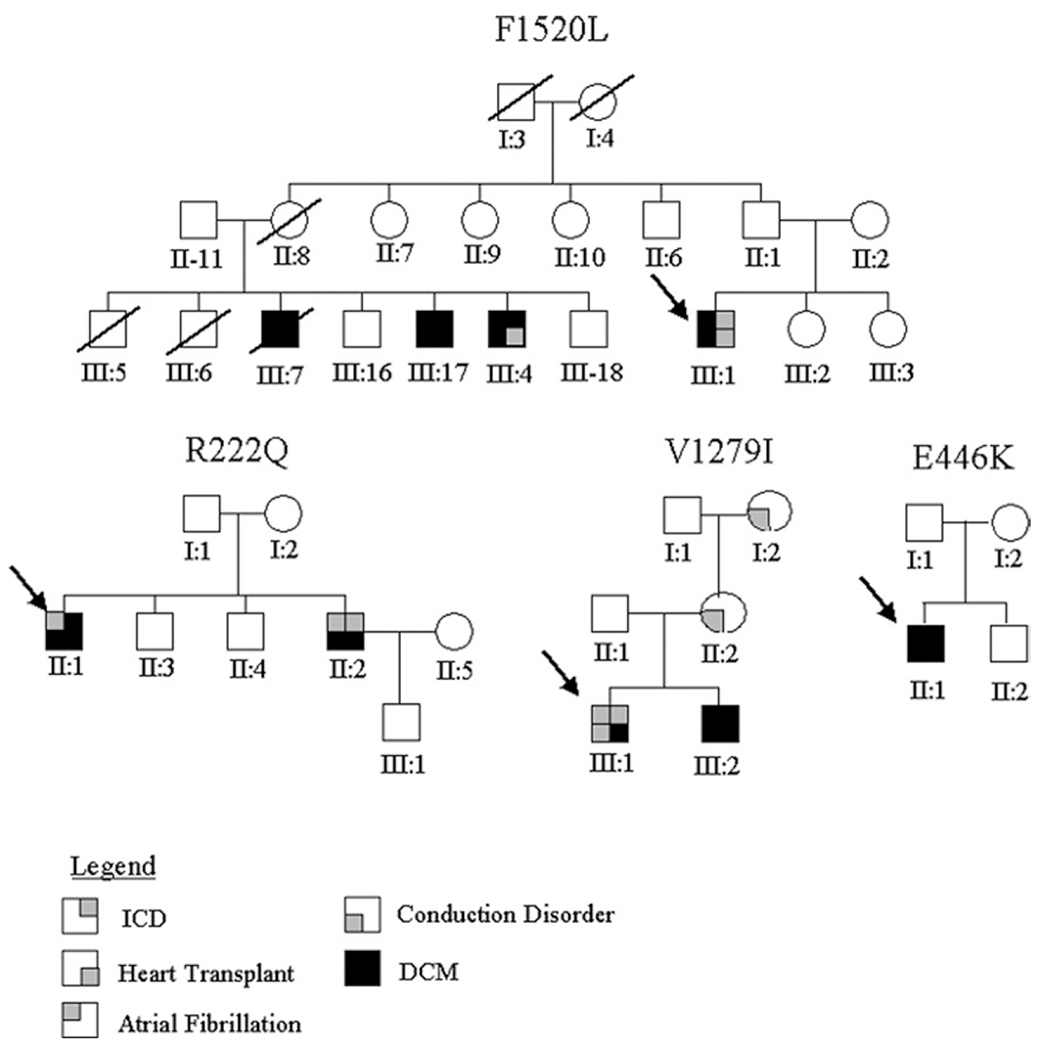
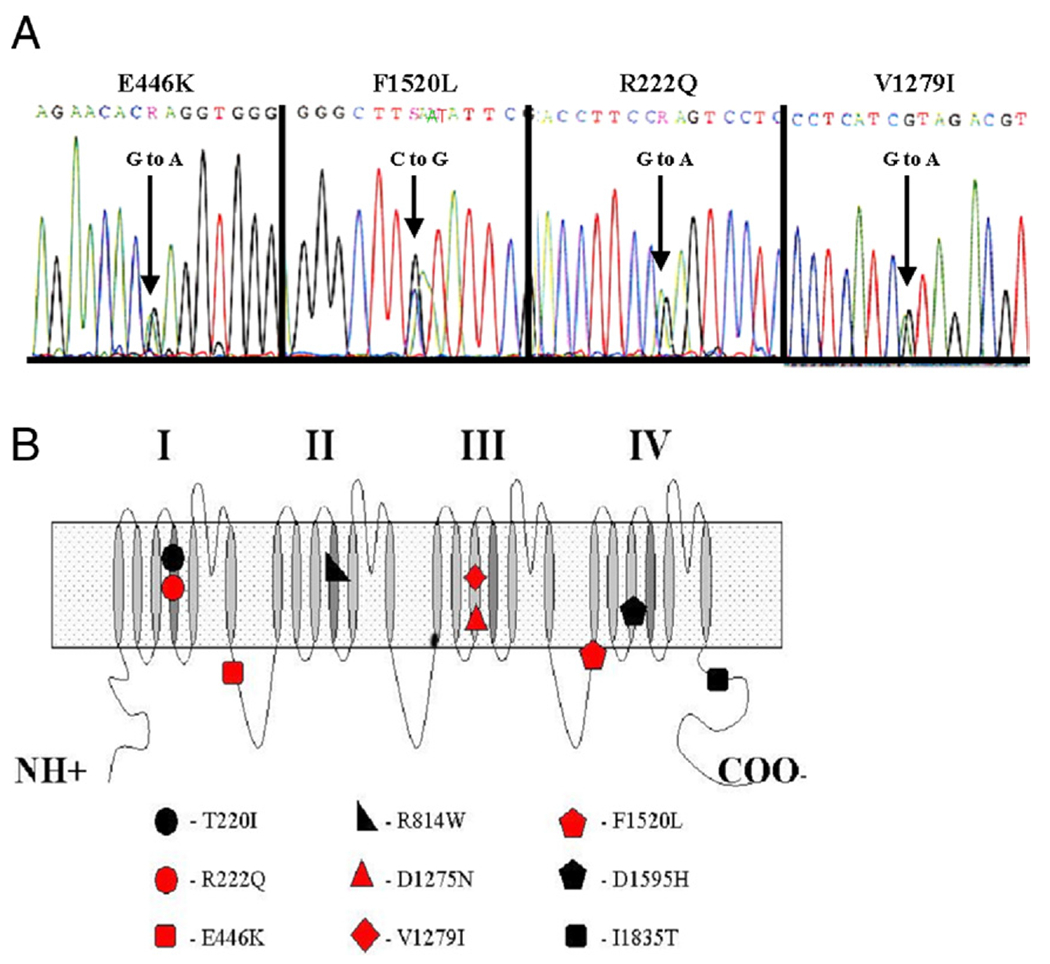
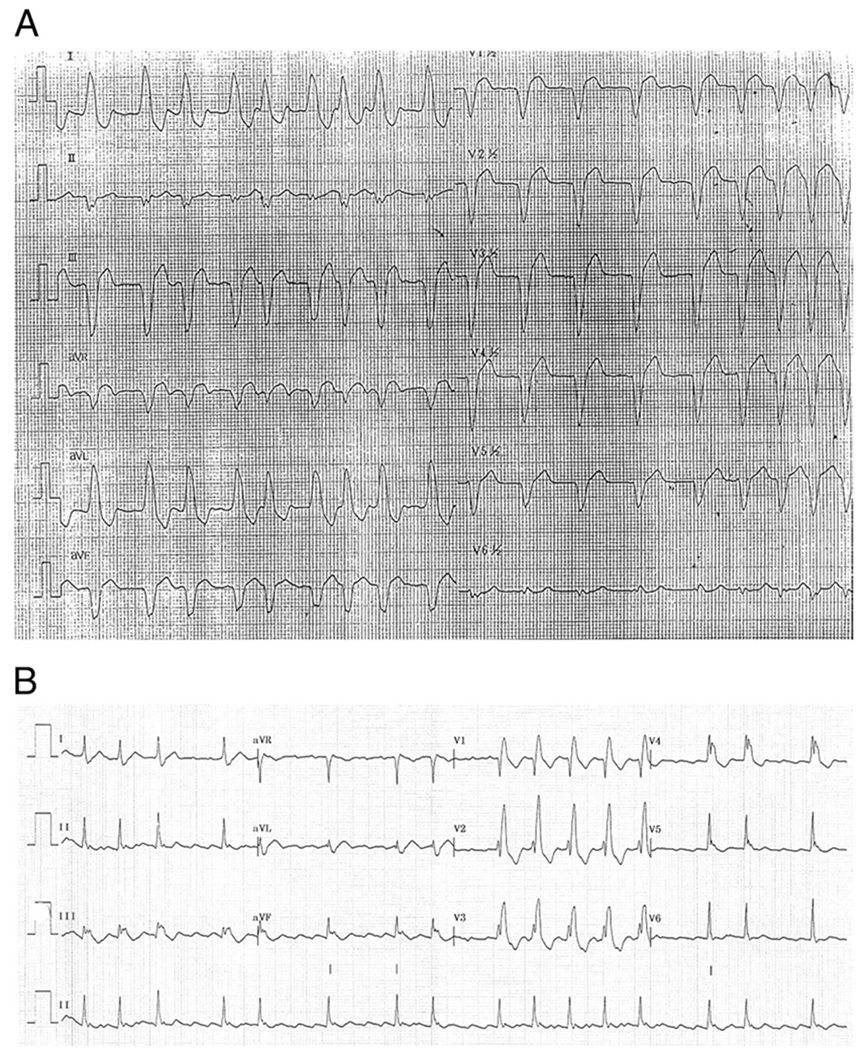
Results: We identified 5 missense SCN5A mutations among our DCM families, including novel mutations E446K, F1520L, and V1279I, as well as previously reported mutations D1275N and R222Q. Of 15 SCN5A mutation carriers in our study, 14 (93%) manifested arrhythmia: supraventricular arrhythmia (13 of 15), including sick sinus syndrome (5 of 15) and atrial fibrillation (9 of 15), ventricular tachycardia (5 of 15), and conduction disease (9 of 15).
Conclusions: Mutations in SCN5A were detected in 1.7% of DCM families. Two-thirds (6 of 9) of all reported DCM mutations in SCN5A localize to the highly conserved homologous S3 and S4 transmembrane segments, suggesting a shared mechanism of disruption of the voltage-sensing mechanism of this channel leading to DCM. Not surprisingly, SCN5A mutation carriers show a strong arrhythmic pattern that has clinical and diagnostic implications.
2011 American College of Cardiology Foundation. Published by Elsevier Inc. All rights reserved.
Figures
Comment in
-
Arrhythmias and dilated cardiomyopathy common pathogenetic pathways?J Am Coll Cardiol. 2011 May 24;57(21):2169-71. doi: 10.1016/j.jacc.2010.11.061. J Am Coll Cardiol. 2011. PMID: 21596232 No abstract available.
References
-
- Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA. Genetic evaluation of cardiomyopathy—a Heart Failure Society of America practice guideline. J Card Fail 2009;15:83–97. - PubMed
-
- Tiso N, Stephan DA, Nava A, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet 2001;10:189–94. - PubMed
-
- Asahi M, Nakayama H, Tada M, Otsu K. Regulation of sarco(endo) plasmic reticulum Ca2+ adenosine triphosphatase by phospholamban and sarcolipin: implication for cardiac hypertrophy and failure. Trends Cardiovasc Med 2003;13:152–7. - PubMed
-
- Schmitt JP, Kamisago M, Asahi M, et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 2003; 299:1410–3. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
