

Epigenetic control of gene expression in the lung
- PMID: 21596832
- PMCID: PMC3114059
- DOI: 10.1164/rccm.201010-1579PP
Epigenetic control of gene expression in the lung
Abstract
Epigenetics is traditionally defined as the study of heritable changes in gene expression caused by mechanisms other than changes in the underlying DNA sequence. There are three main classes of epigenetic marks--DNA methylation, modifications of histone tails, and noncoding RNAs--each of which may be influenced by the environment, diet, diseases, and ageing. Importantly, epigenetic marks have been shown to influence immune cell maturation and are associated with the risk of developing various forms of cancer, including lung cancer. Moreover, there is emerging evidence that these epigenetic marks affect gene expression in the lung and are associated with benign lung diseases, such as asthma, chronic obstructive pulmonary disease, and interstitial lung disease. Technological advances have made it feasible to study epigenetic marks in the lung, and it is anticipated that this knowledge will enhance our understanding of the dynamic biology in the lung and lead to the development of novel diagnostic and therapeutic approaches for our patients with lung disease.
Figures

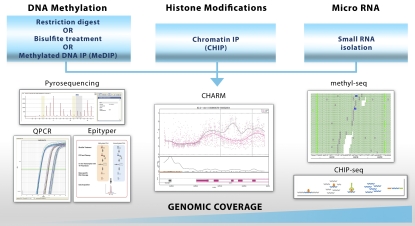
References
-
- Allis CD, Jenuwein T, Reinberg D, editors. Epigenetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2009.
-
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol 1987;196:261–282. - PubMed
-
- Guilleret I, Yan P, Grange F, Braunschweig R, Bosman FT, Benhattar J. Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int J Cancer 2002;101:335–341. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
