

Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases
- PMID: 21597461
- PMCID: PMC3178424
- DOI: 10.1038/cdd.2011.65
Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases
Abstract
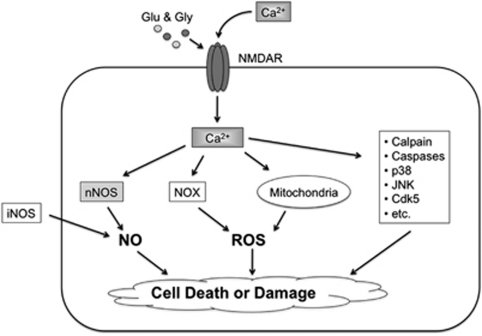
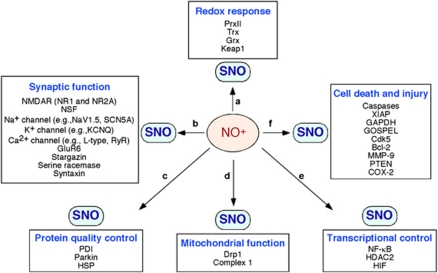
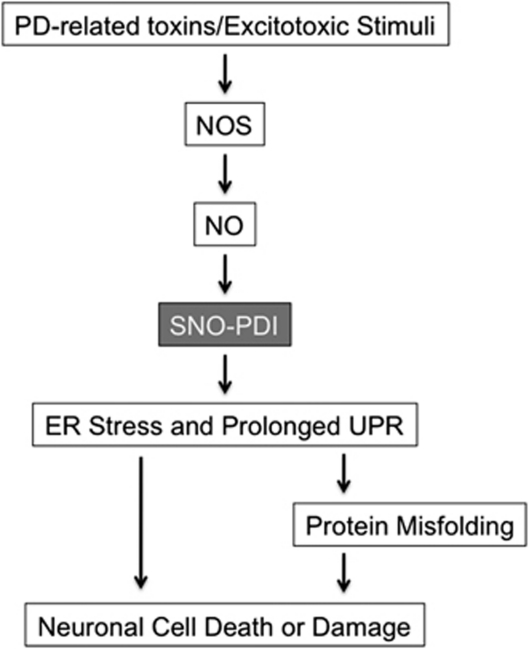
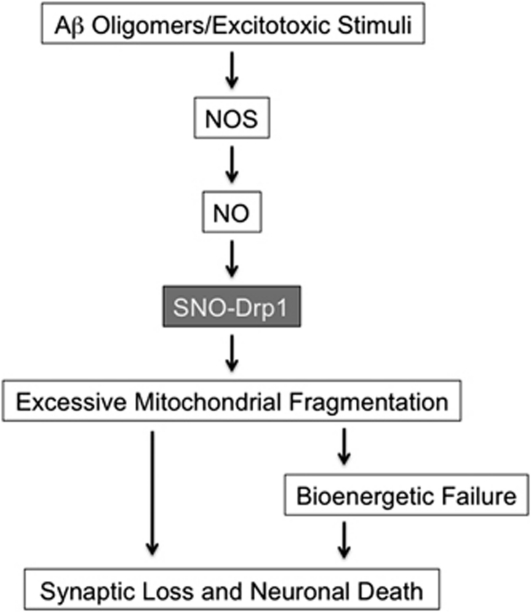
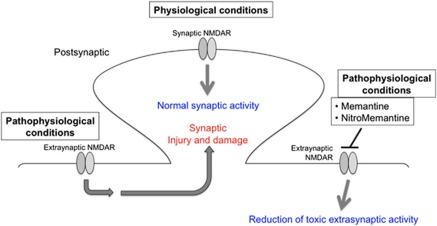
The pathological processes of neurodegenerative disorders such as Alzheimer's and Parkinson's diseases engender synaptic and neuronal cell damage. While mild oxidative and nitrosative (nitric oxide (NO)-related) stress mediates normal neuronal signaling, excessive accumulation of these free radicals is linked to neuronal cell injury or death. In neurons, N-methyl-D-aspartate (NMDA) receptor (NMDAR) activation and subsequent Ca(2+) influx can induce the generation of NO via neuronal NO synthase. Emerging evidence has demonstrated that S-nitrosylation, representing covalent reaction of an NO group with a critical protein thiol, mediates the vast majority of NO signaling. Analogous to phosphorylation and other posttranslational modifications, S-nitrosylation can regulate the biological activity of many proteins. Here, we discuss recent studies that implicate neuropathogenic roles of S-nitrosylation in protein misfolding, mitochondrial dysfunction, synaptic injury, and eventual neuronal loss. Among a growing number of S-nitrosylated proteins that contribute to disease pathogenesis, in this review we focus on S-nitrosylated protein-disulfide isomerase (forming SNO-PDI) and dynamin-related protein 1 (forming SNO-Drp1). Furthermore, we describe drugs, such as memantine and newer derivatives of this compound that can prevent both hyperactivation of extrasynaptic NMDARs as well as downstream pathways that lead to nitrosative stress, synaptic damage, and neuronal loss.
Figures
References
-
- Beal MF. Experimental models of Parkinson's disease. Nat Rev Neurosci. 2001;2:325–334. - PubMed
-
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. - PubMed
-
- Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5:160–170. - PubMed
-
- Garthwaite J, Charles SL, Chess-Williams R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature. 1988;336:385–388. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
