

Dense deposit disease
- PMID: 21601923
- PMCID: PMC3142282
- DOI: 10.1016/j.molimm.2011.04.005
Dense deposit disease
Abstract
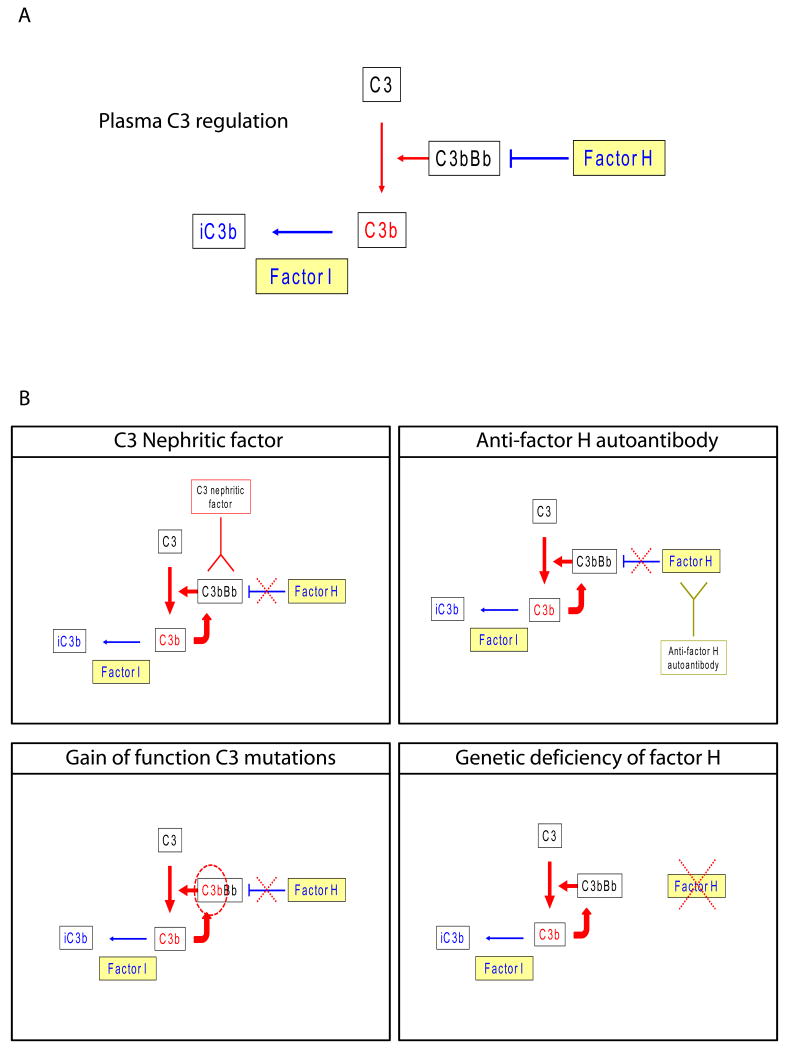
Dense deposit disease (DDD) is an orphan disease that primarily affects children and young adults without sexual predilection. Studies of its pathophysiology have shown conclusively that it is caused by fluid-phase dysregulation of the alternative pathway of complement, however the role played by genetics and autoantibodies like C3 nephritic factors must be more thoroughly defined if we are to make an impact in the clinical management of this disease. There are currently no mechanism-directed therapies to offer affected patients, half of whom progress to end stage renal failure disease within 10 years of diagnosis. Transplant recipients face the dim prospect of disease recurrence in their allografts, half of which ultimately fail. More detailed genetic and complement studies of DDD patients may make it possible to identify protective factors prognostic for naïve kidney and transplant survival, or conversely risk factors associated with progression to renal failure and allograft loss. The pathophysiology of DDD suggests that a number of different treatments warrant consideration. As advances are made in these areas, there will be a need to increase healthcare provider awareness of DDD by making resources available to clinicians to optimize care for DDD patients.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
References
-
- Appel GB, Cook T, Hageman G, Jennette C, Kashgarian M, Kirschfink M, Lambris JD, Lanning L, Lutz HU, Meri S, Rose NR, Salant DJ, Sethi S, Smith RJH, Smoyer W, Tully HF, Tully SP, Walker P, Welsh M, Würzner R, Zipfel PF. Membranoproliferative glomerulonephritis type II (Dense Deposit Disease): An update. J Am Soc Nephrol. 2005;16:1392–1403. - PubMed
-
- Bircan Z, Toprak D, Kilicaslan I, Solakoglu S, Uysal V, Ponard D, Turker G. Factor H deficiency and fibrillary glomerulopathy. Nephrol Dial Transplant. 2004;19:727–730. - PubMed
-
- Botto M, Kirschfink M, Macor P, Pickering MC, Würzner R, Tedesco F. Complement in human diseases: Lessons from complement deficiencies. Mol Immunol. 2009;46:2774–83. - PubMed
-
- Braun MC, Stablein DM, Hamiwka LA, Bell L, Bartosh SM, Strife CF. Recurrence of membranoproliferative glomerulonephritis type II in renal allografts: The north american pediatric renal transplant cooperative study experience. JASN. 2005;16:2225–2233. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
