

Expanded methyl-sensitive cut counting reveals hypomethylation as an epigenetic state that highlights functional sequences of the genome
- PMID: 21602498
- PMCID: PMC3111303
- DOI: 10.1073/pnas.1105713108
Expanded methyl-sensitive cut counting reveals hypomethylation as an epigenetic state that highlights functional sequences of the genome
Erratum in
- Proc Natl Acad Sci U S A. 2013 Mar 19;110(12):4853
Abstract
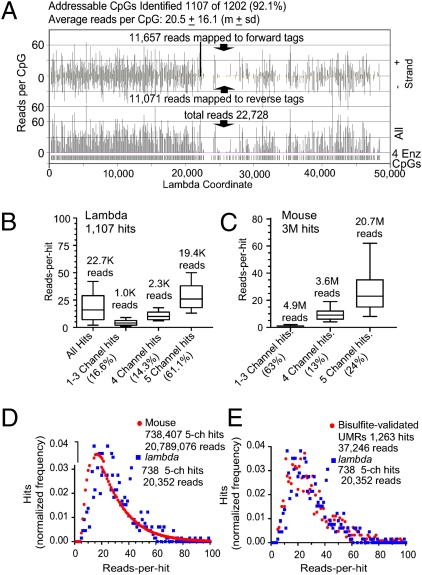
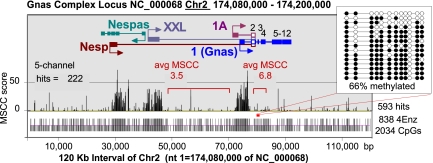
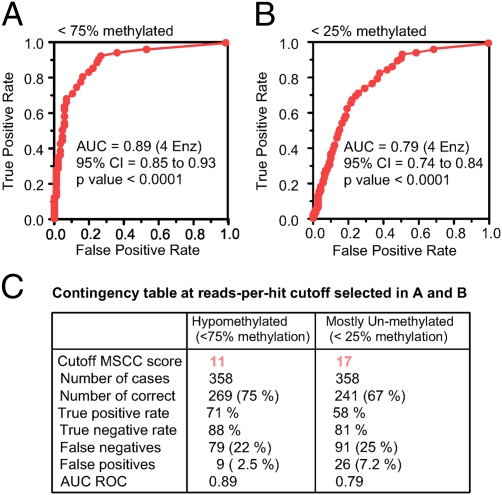
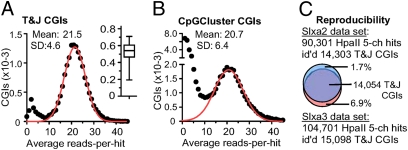
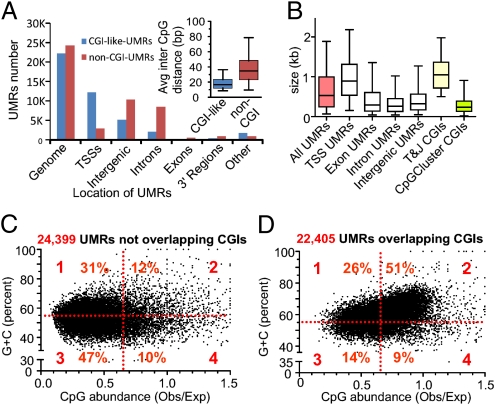
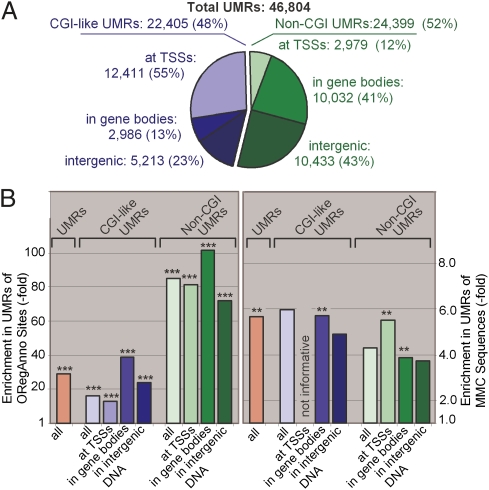
Methyl-sensitive cut counting (MSCC) with the HpaII methylation-sensitive restriction enzyme is a cost-effective method to pinpoint unmethylated CpGs at single base-pair resolution. However, it has the drawback of addressing only CpGs in the context of the CCGG site, leaving out the remainder of the possible 16 XCGX tetranucleotides in which CpGs are found. We expanded MSCC to include three additional enzymes to address a total of 5 of the 16 XCGX combinations. This allowed us to survey methylation at about one-third of all a mammalian genome's CpGs. Applied to mouse liver DNA, we correctly confirmed data reported with other methods showing hypomethylation to be concentrated at promoters and in CpG islands (CGIs), with gene bodies and intergenic regions being mostly methylated. Grouping unmethylated CpGs, characterized by high MSCC scores (7% false discovery rate), we found a large number of unmethylated regions not qualifying as CGIs located in intergenic and intronic regions, which are highly enriched in functional DNA sequences (open regulatory annotation database) as well as in noncoding yet highly conserved mammalian sequences thought to be important but with as yet unknown function. About 50% of MSCC-defined unmethylated regions do not overlap algorithm-defined CGIs and offer a novel search space in which new functionalities of DNA may be found in health and disease.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Suzuki MM, Bird A. DNA methylation landscapes: Provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. - PubMed
-
- Leonhardt H, Bestor TH. Structure, function and regulation of mammalian DNA methyltransferase. EXS. 1993;64:109–119. - PubMed
-
- Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
