

De novo enzyme design using Rosetta3
- PMID: 21603656
- PMCID: PMC3095599
- DOI: 10.1371/journal.pone.0019230
De novo enzyme design using Rosetta3
Abstract
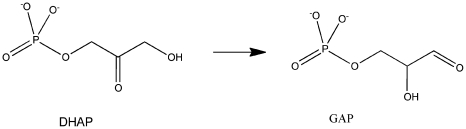
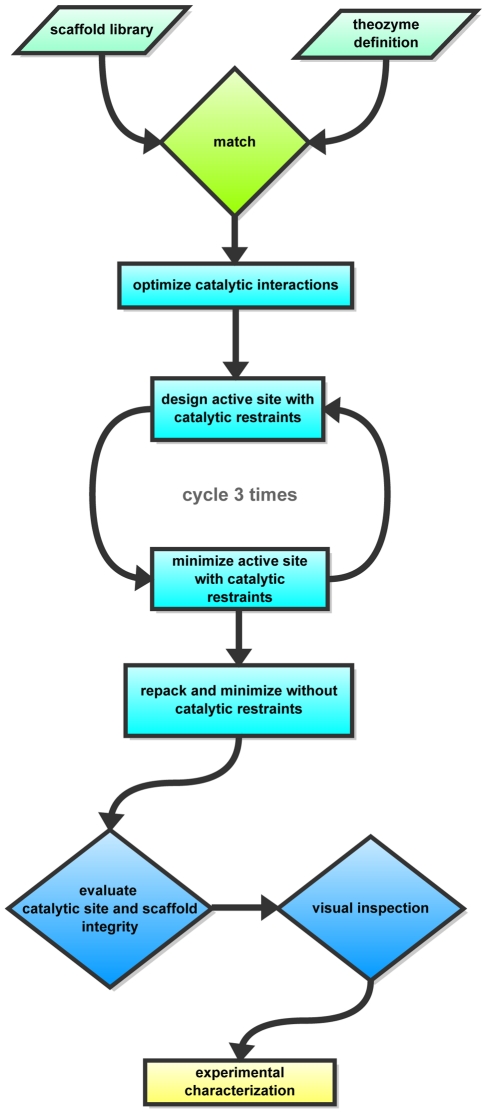
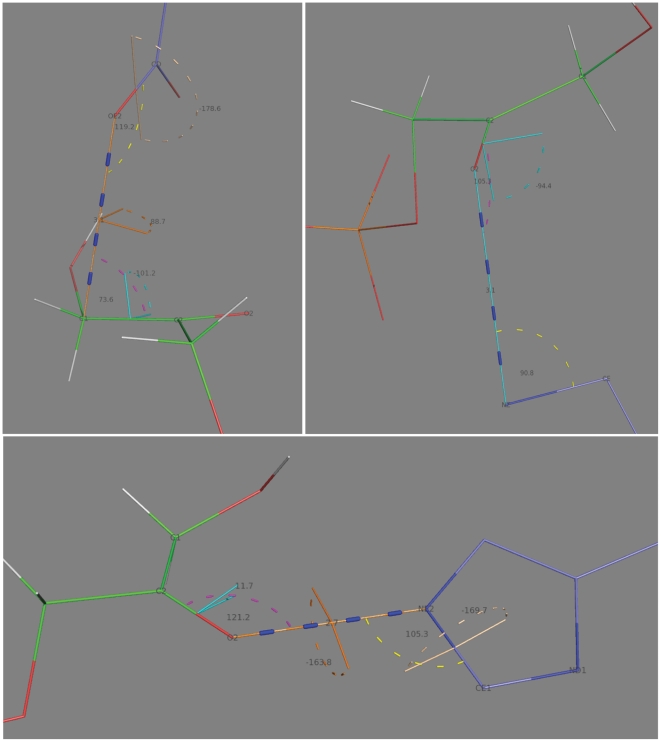
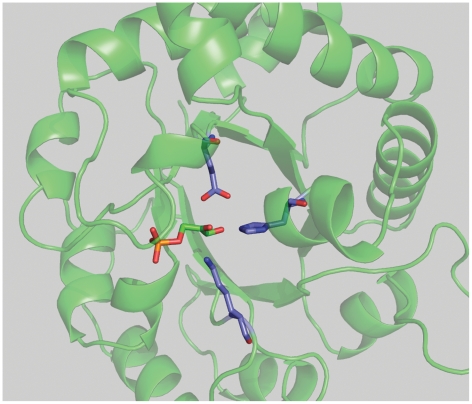
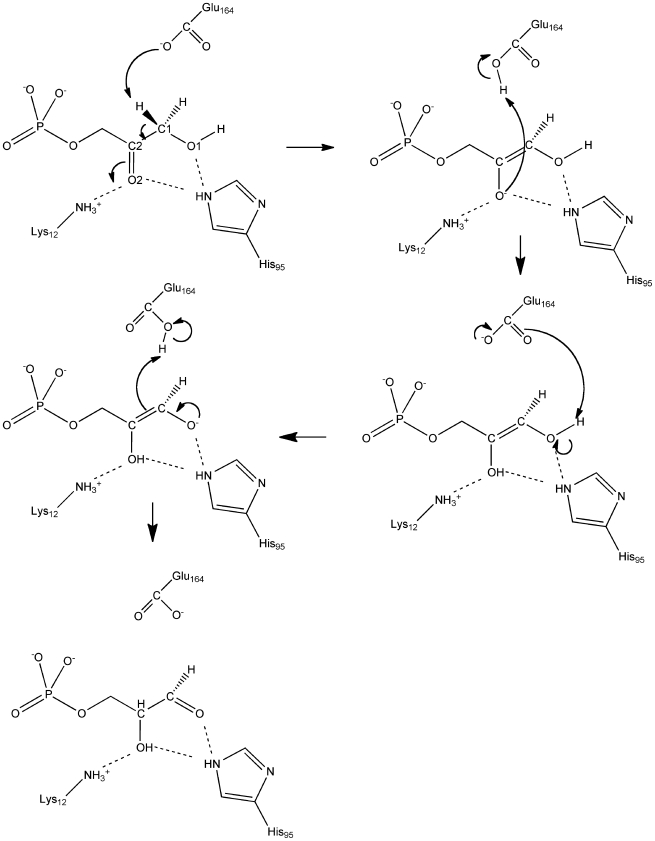
The Rosetta de novo enzyme design protocol has been used to design enzyme catalysts for a variety of chemical reactions, and in principle can be applied to any arbitrary chemical reaction of interest. The process has four stages: 1) choice of a catalytic mechanism and corresponding minimal model active site, 2) identification of sites in a set of scaffold proteins where this minimal active site can be realized, 3) optimization of the identities of the surrounding residues for stabilizing interactions with the transition state and primary catalytic residues, and 4) evaluation and ranking the resulting designed sequences. Stages two through four of this process can be carried out with the Rosetta package, while stage one needs to be done externally. Here, we demonstrate how to carry out the Rosetta enzyme design protocol from start to end in detail using for illustration the triosephosphate isomerase reaction.
Conflict of interest statement
Figures
References
-
- Röthlisberger Daniela, et al. “Kemp elimination catalysts by computational enzyme design,”. Nature. 2008;453, no.7192:190–195. - PubMed
-
- Garcia-Viloca Mireia, et al. “How enzymes work: analysis by modern rate theory and computer simulations,”. Science. 2004;303, no.5655:186–195. (New York, N.Y.) - PubMed
-
- Tantillo DJ, Chen J, Houk KN. “Theozymes and compuzymes: theoretical models for biological catalysis,” . Current Opinion in Chemical Biology. 1998;2, no.6:743–750. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
