

Therapeutics development in myotonic dystrophy type 1
- PMID: 21607985
- PMCID: PMC3136655
- DOI: 10.1002/mus.22090
Therapeutics development in myotonic dystrophy type 1
Abstract
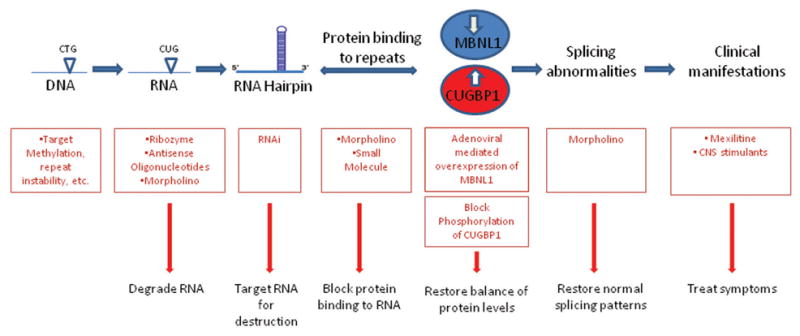
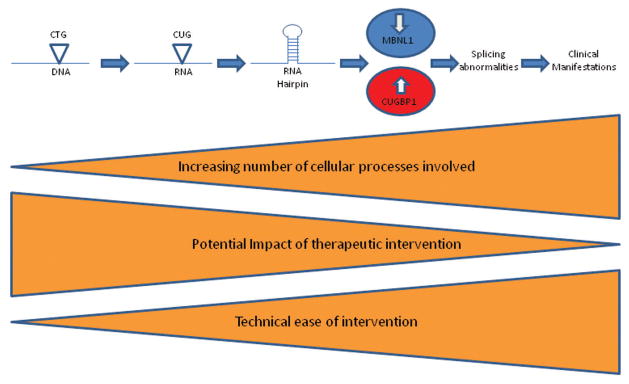
Myotonic dystrophy (DM1), the most common adult muscular dystrophy, is a multisystem, autosomal dominant genetic disorder caused by an expanded CTG repeat that leads to nuclear retention of a mutant RNA and subsequent RNA toxicity. Significant insights into the molecular mechanisms of RNA toxicity have led to the previously unforeseen possibility that treating DM1 is a viable prospect. In this review, we briefly present the clinical picture in DM1, and describe how the research in understanding the pathogenesis of RNA toxicity in DM1 has led to targeted approaches to therapeutic development at various steps in the pathogenesis of the disease. We discuss the promise and current limitations of each with an emphasis on RNA-based therapeutics and small molecules. We conclude with a discussion of the unmet need for clinical tools and outcome measures that are essential prerequisites to proceed in evaluating these potential therapies in clinical trials.
Copyright © 2011 Wiley Periodicals, Inc.
Figures
References
-
- Harper PS. Myotonic Dystrophy. London: W.B. Saunders; 1989.
-
- Emery AE. Population frequencies of inherited neuromuscular diseases--a world survey. Neuromuscul Disord. 1991;1(1):19–29. - PubMed
-
- Yotova V, Labuda D, Zietkiewicz E, Gehl D, Lovell A, Lefebvre JF, Bourgeois S, Lemieux-Blanchard E, Labuda M, Vezina H, Houde L, Tremblay M, Toupance B, Heyer E, Hudson TJ, Laberge C. Anatomy of a founder effect: myotonic dystrophy in Northeastern Quebec. Hum Genet. 2005;117(2–3):177–187. - PubMed
-
- Griggs R, Miller RG, Mendell JR. Evaluation and Treatment of Myopathies. Philadelphia: F.A. Davis Company; 1995.
-
- de Die-Smulders CE, Howeler CJ, Thijs C, Mirandolle JF, Anten HB, Smeets HJ, Chandler KE, Geraedts JP. Age and causes of death in adult-onset myotonic dystrophy. Brain. 1998;121 ( Pt 8):1557–1563. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
