

HMM-FRAME: accurate protein domain classification for metagenomic sequences containing frameshift errors
- PMID: 21609463
- PMCID: PMC3115854
- DOI: 10.1186/1471-2105-12-198
HMM-FRAME: accurate protein domain classification for metagenomic sequences containing frameshift errors
Abstract
Background: Protein domain classification is an important step in metagenomic annotation. The state-of-the-art method for protein domain classification is profile HMM-based alignment. However, the relatively high rates of insertions and deletions in homopolymer regions of pyrosequencing reads create frameshifts, causing conventional profile HMM alignment tools to generate alignments with marginal scores. This makes error-containing gene fragments unclassifiable with conventional tools. Thus, there is a need for an accurate domain classification tool that can detect and correct sequencing errors.
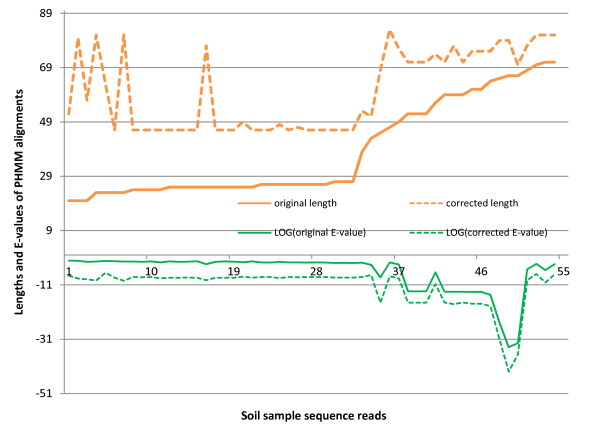
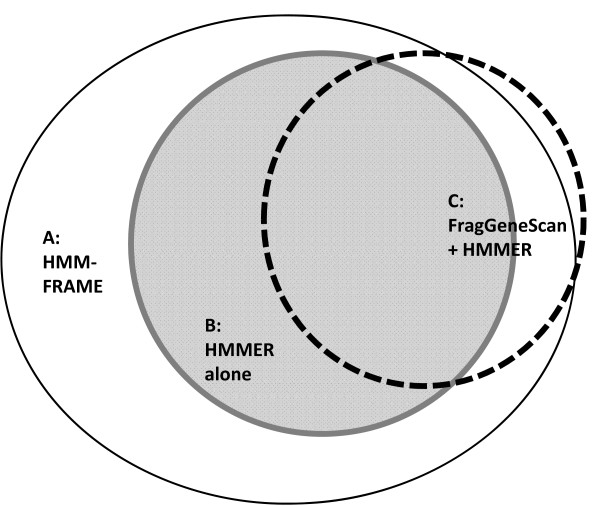
Results: We introduce HMM-FRAME, a protein domain classification tool based on an augmented Viterbi algorithm that can incorporate error models from different sequencing platforms. HMM-FRAME corrects sequencing errors and classifies putative gene fragments into domain families. It achieved high error detection sensitivity and specificity in a data set with annotated errors. We applied HMM-FRAME in Targeted Metagenomics and a published metagenomic data set. The results showed that our tool can correct frameshifts in error-containing sequences, generate much longer alignments with significantly smaller E-values, and classify more sequences into their native families.
Conclusions: HMM-FRAME provides a complementary protein domain classification tool to conventional profile HMM-based methods for data sets containing frameshifts. Its current implementation is best used for small-scale metagenomic data sets. The source code of HMM-FRAME can be downloaded at http://www.cse.msu.edu/~zhangy72/hmmframe/ and at https://sourceforge.net/projects/hmm-frame/.
Figures

 is the jth amino acid of a peptide sequence derived under reading frame i. The correct peptide sequence can be derived from the error-free sequence (shown on the top of the figure) under reading frame 1. Because of insertions of two nucleotides (bolded X and Y), the correct peptide sequence is the concatenation of three short peptide sequences derived using different reading frames. Thus, each peptide sequence derived using one reading frame can only generate short alignments with insignificant scores.
is the jth amino acid of a peptide sequence derived under reading frame i. The correct peptide sequence can be derived from the error-free sequence (shown on the top of the figure) under reading frame 1. Because of insertions of two nucleotides (bolded X and Y), the correct peptide sequence is the concatenation of three short peptide sequences derived using different reading frames. Thus, each peptide sequence derived using one reading frame can only generate short alignments with insignificant scores.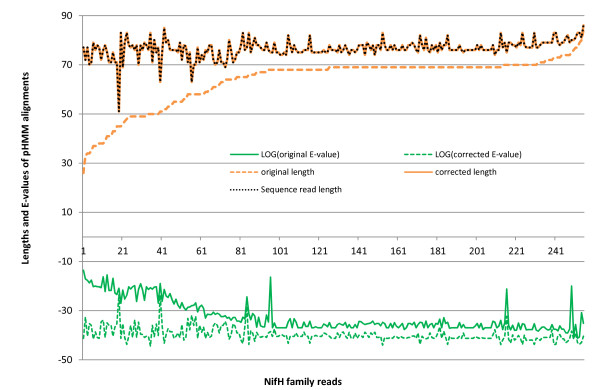
References
-
- HMMER3: a new generation of sequence homology search software. http://hmmer.janelia.org/
-
- Guan X, Uberbacher E. Alignments of DNA and protein sequences containing frameshift errors. Comput Appl Biosci. 1996;12:31–40. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
