

Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy
- PMID: 21613495
- PMCID: PMC3351137
- DOI: 10.1523/JNEUROSCI.6412-10.2011
Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy
Abstract
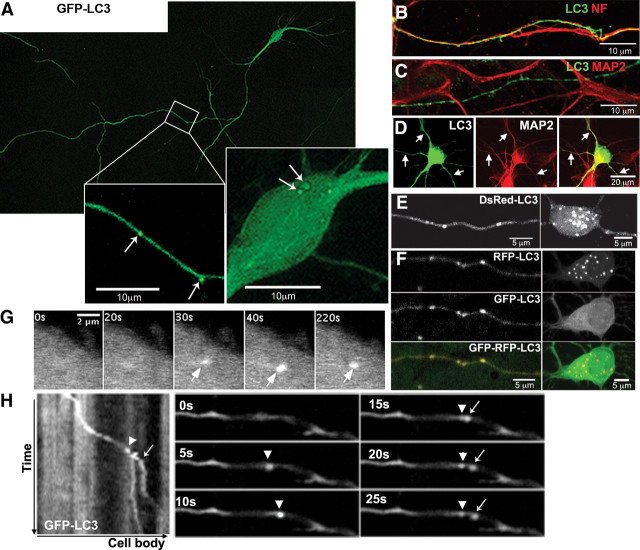
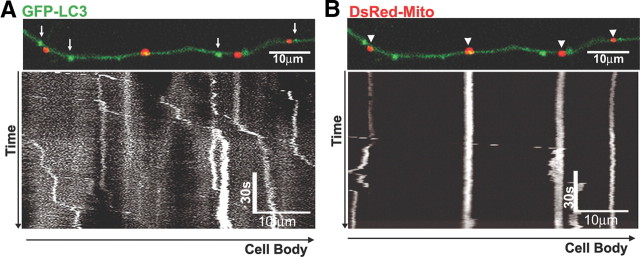
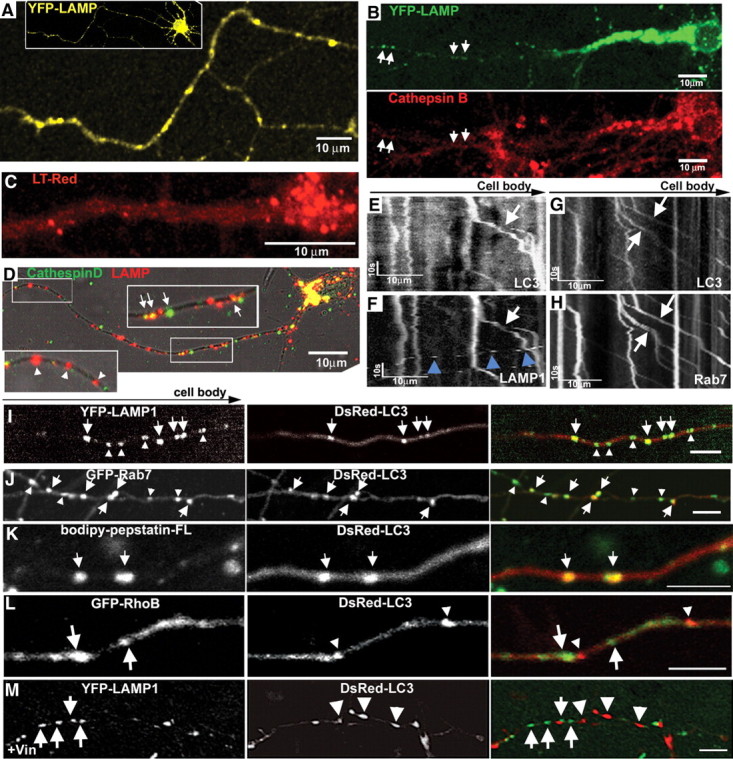
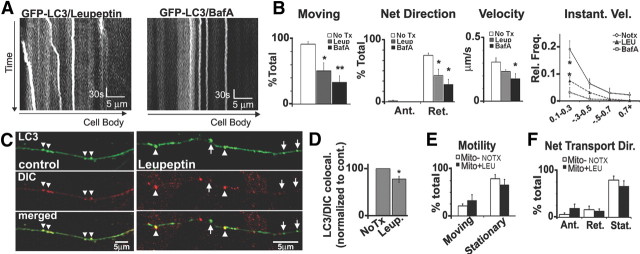
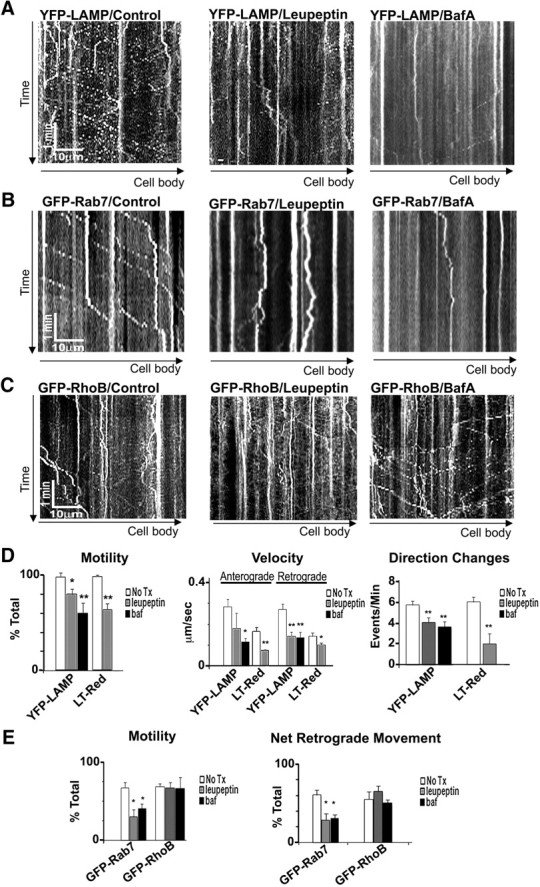
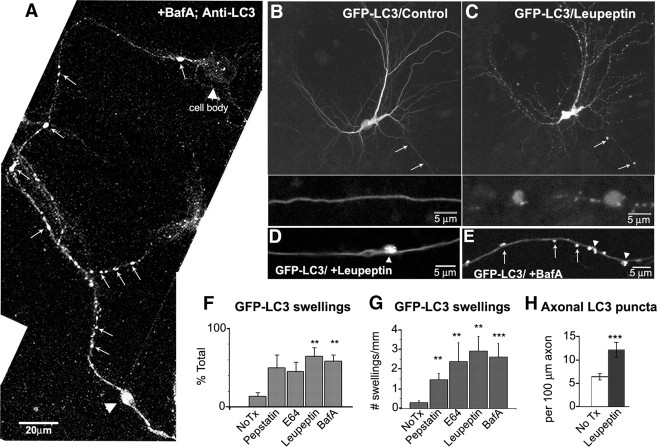
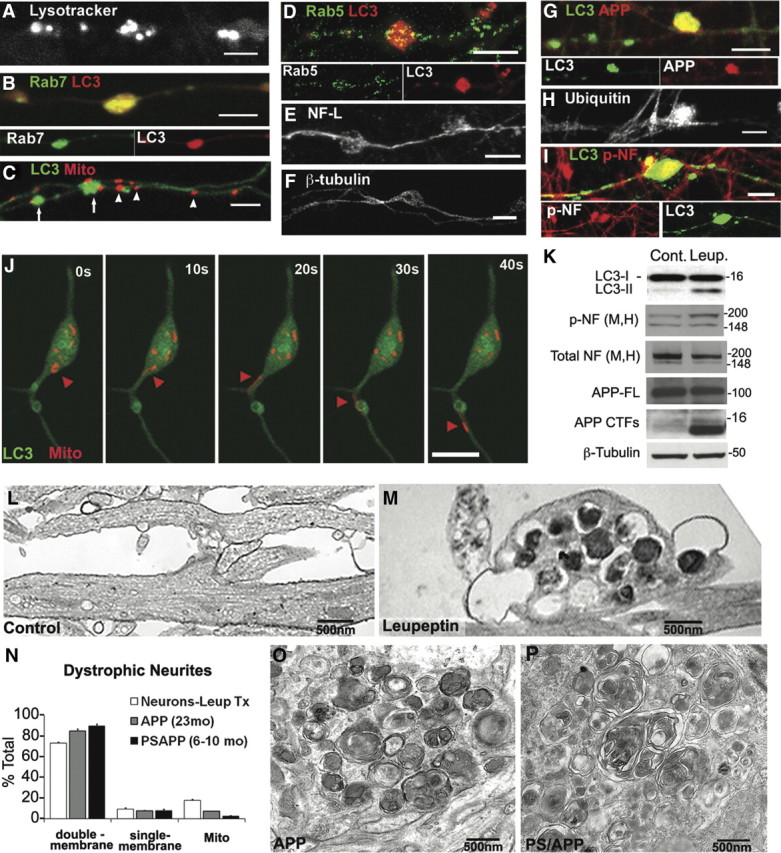
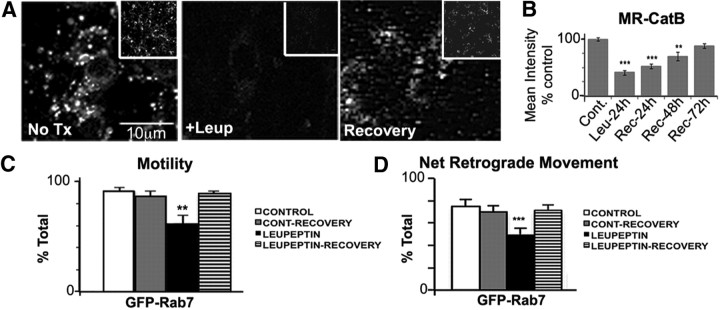
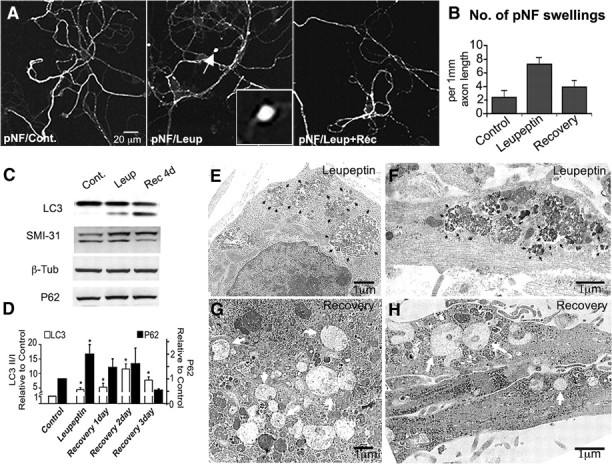
In the hallmark neuritic dystrophy of Alzheimer's disease (AD), autophagic vacuoles containing incompletely digested proteins selectively accumulate in focal axonal swellings, reflecting defects in both axonal transport and autophagy. Here, we investigated the possibility that impaired lysosomal proteolysis could be a basis for both of these defects leading to neuritic dystrophy. In living primary mouse cortical neurons expressing fluorescence-tagged markers, LC3-positive autophagosomes forming in axons rapidly acquired the endo-lysosomal markers Rab7 and LAMP1 and underwent exclusive retrograde movement. Proteolytic clearance of these transported autophagic vacuoles was initiated after fusion with bidirectionally moving lysosomes that increase in number at more proximal axon levels and in the perikaryon. Disrupting lysosomal proteolysis by either inhibiting cathepsins directly or by suppressing lysosomal acidification slowed the axonal transport of autolysosomes, late endosomes, and lysosomes and caused their selective accumulation within dystrophic axonal swellings. Mitochondria and other organelles lacking cathepsins moved normally under these conditions, indicating that the general functioning of the axonal transport system was preserved. Dystrophic swellings induced by lysosomal proteolysis inhibition resembled in composition those in several mouse models of AD and also acquired other AD-like features, including immunopositivity for ubiquitin, amyloid precursor protein, and hyperphosphorylated neurofilament proteins. Restoration of lysosomal proteolysis reversed the affected movements of proteolytic Rab7 vesicles, which in turn essentially cleared autophagic substrates and reversed the axonal dystrophy. These studies identify the AD-associated defects in neuronal lysosomal proteolysis as a possible basis for the selective transport abnormalities and highly characteristic pattern of neuritic dystrophy associated with AD.
Figures
References
-
- Adalbert R, Nogradi A, Babetto E, Janeckova L, Walker SA, Kerschensteiner M, Misgeld T, Coleman MP. Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain. 2009;132:402–416. - PubMed
-
- Bahr BA, Bendiske J. The neuropathogenic contributions of lysosomal dysfunction. J Neurochem. 2002;83:481–489. - PubMed
-
- Ballabio A, Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta. 2009;1793:684–696. - PubMed
-
- Bampton ETW, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dymanics of autophagy visualizes in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous