

Endogenous muscle atrophy F-box mediates pressure overload-induced cardiac hypertrophy through regulation of nuclear factor-kappaB
- PMID: 21617130
- PMCID: PMC3257317
- DOI: 10.1161/CIRCRESAHA.110.238717
Endogenous muscle atrophy F-box mediates pressure overload-induced cardiac hypertrophy through regulation of nuclear factor-kappaB
Abstract
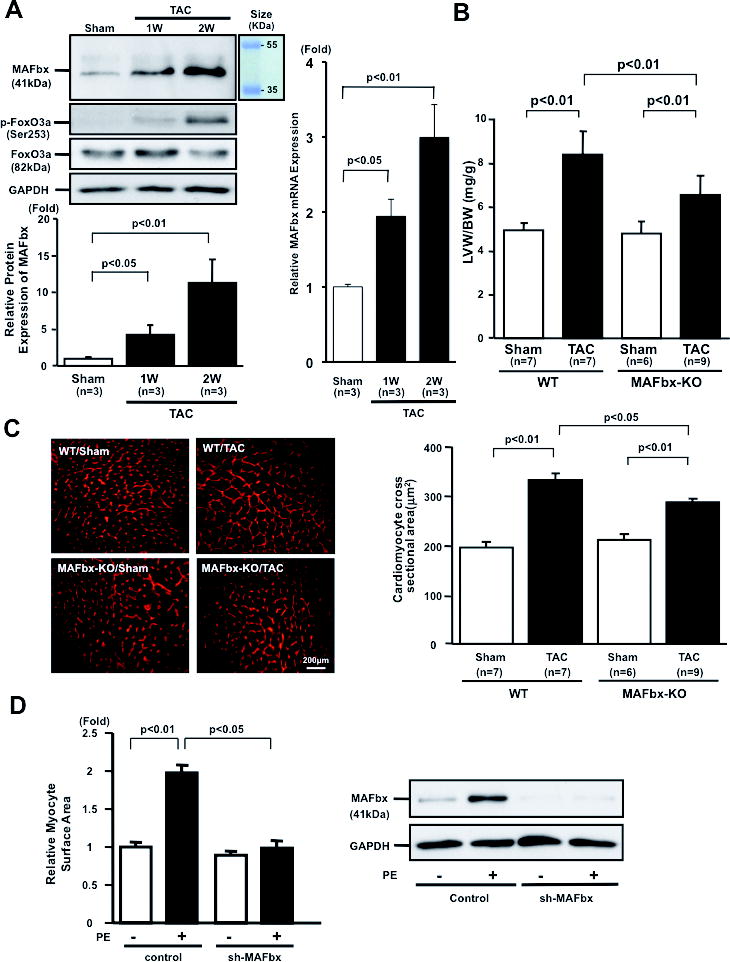
Rationale: Overexpression of muscle atrophy F-box (MAFbx/atrogin-1), an E3 ubiquitin ligase, induces proteasomal degradation in cardiomyocytes. The role of endogenous MAFbx in regulating cardiac hypertrophy and failure remains unclear.
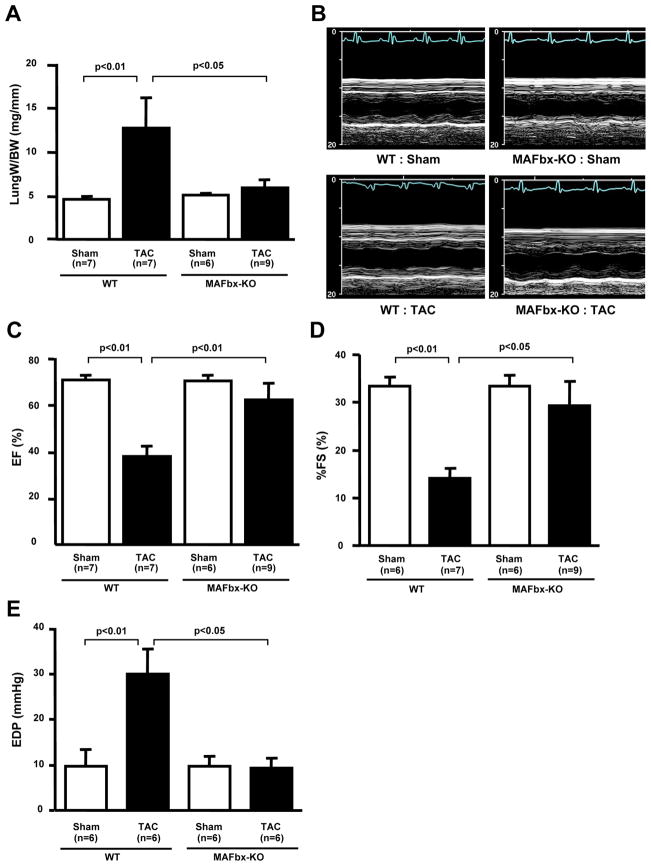
Objective: We investigated the role of MAFbx in regulating cardiac hypertrophy and function in response to pressure overload. Transverse aortic constriction (TAC) was applied to MAFbx knockout (KO) and wild-type (WT) mice.
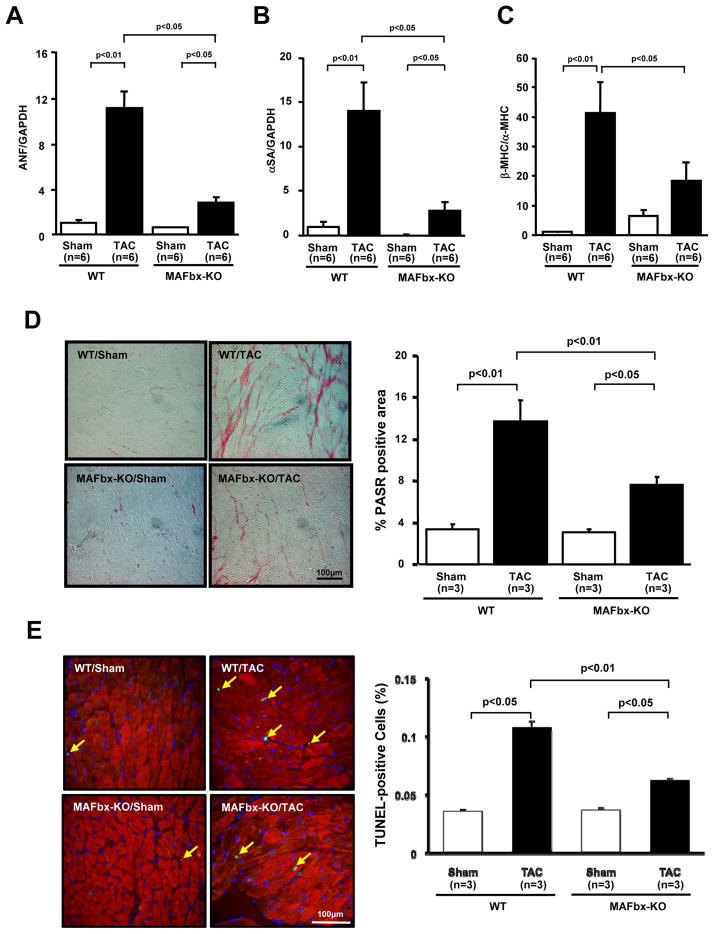
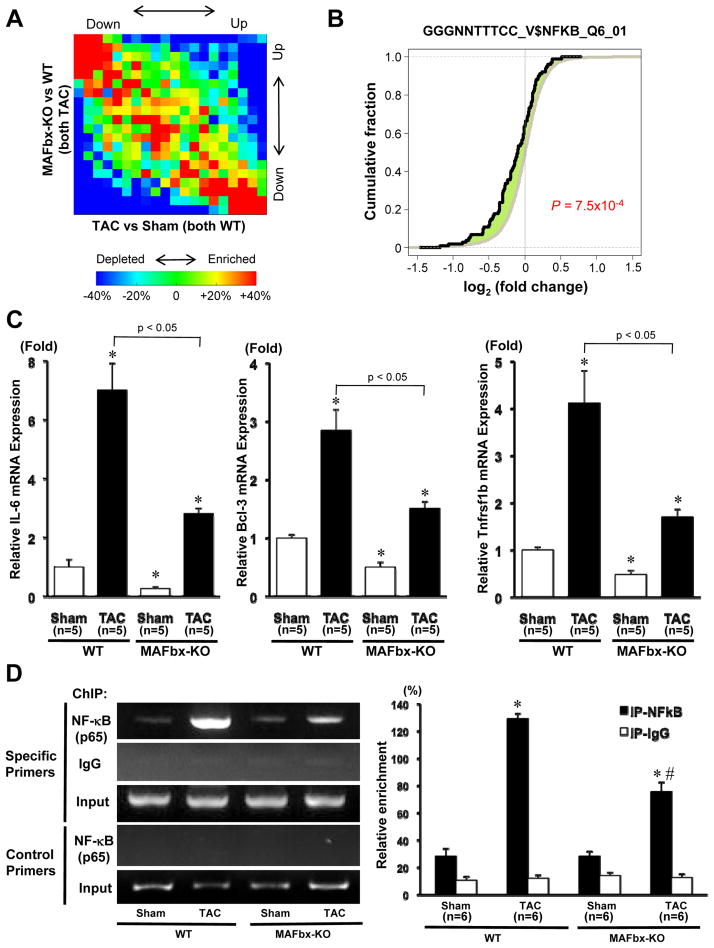
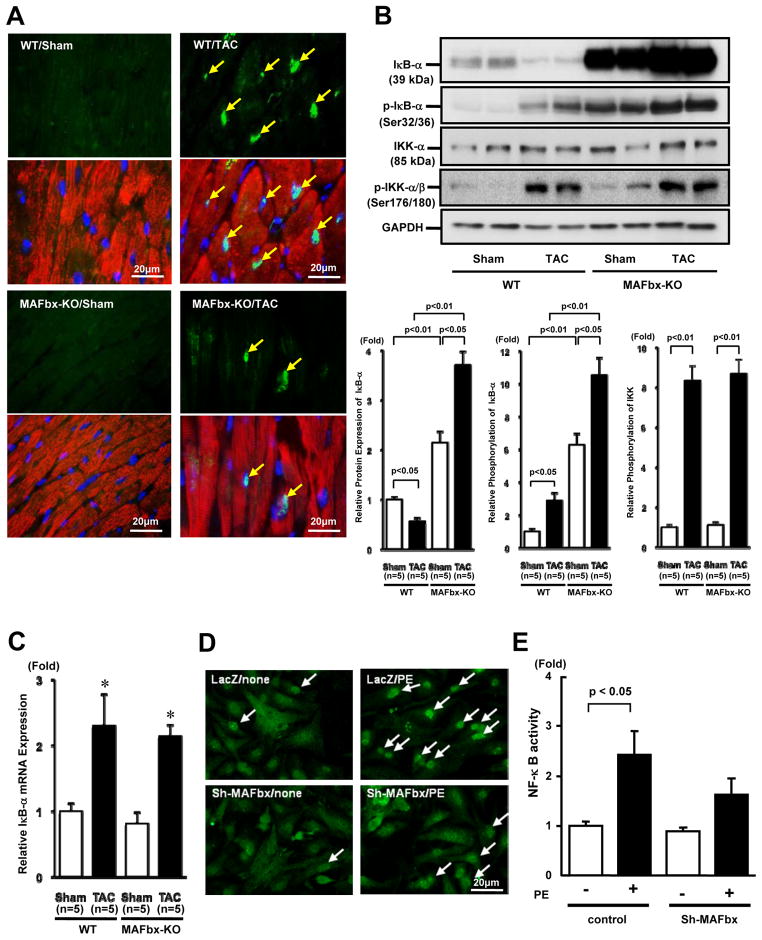
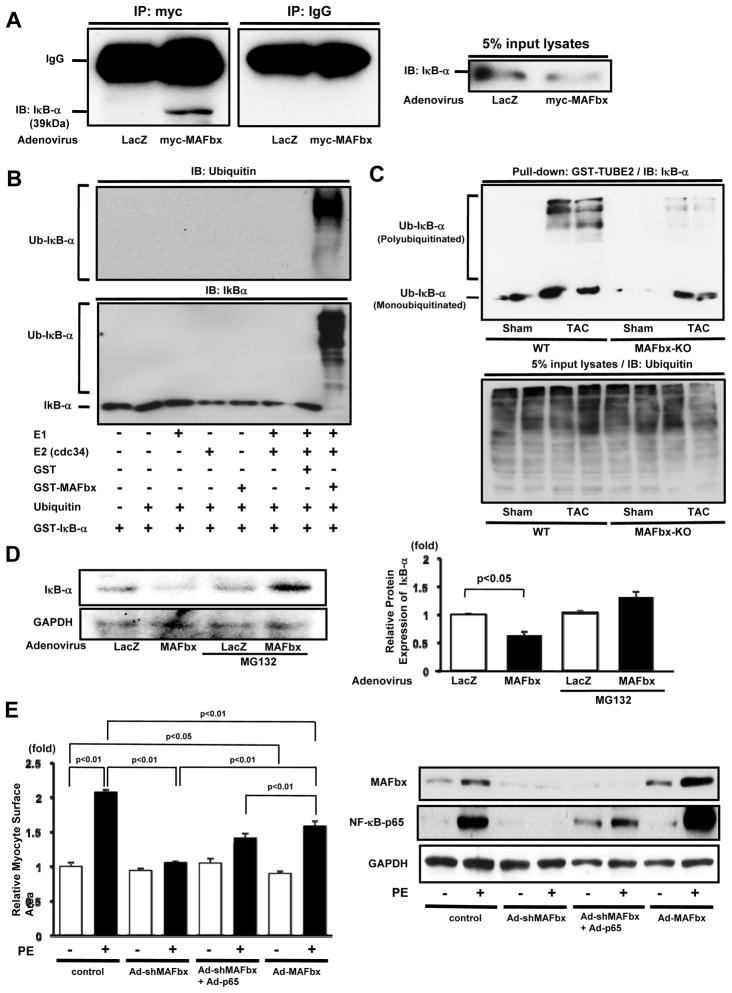
Methods and results: Expression of MAFbx in WT mice was significantly increased by TAC. TAC-induced increases in cardiac hypertrophy were significantly smaller in MAFbx KO than in WT mice. There was significantly less lung congestion and interstitial fibrosis in MAFbx KO than in WT mice. MAFbx KO also inhibited β-adrenergic cardiac hypertrophy. DNA microarray analysis revealed that activation of genes associated with the transcription factor binding site for the nuclear factor-κB family were inhibited in MAFbx KO mice compared with WT mice after TAC. Although the levels of IκB-α were significantly decreased after TAC in WT mice, they were increased in MAFbx KO mice. MAFbx regulates ubiquitination and proteasomal degradation of IκB-α in cardiomyocytes. In primary cultured rat cardiomyocytes, phenylephrine-induced activation of nuclear factor-κB and hypertrophy were significantly suppressed by MAFbx knockdown but were partially rescued by overexpression of nuclear factor-κB p65.
Conclusions: MAFbx plays an essential role in mediating cardiac hypertrophy in response to pressure overload. Downregulation of MAFbx inhibits cardiac hypertrophy in part through stabilization of IκB-α and inactivation of nuclear factor-κB. Taken together, inhibition of MAFbx attenuates pathological hypertrophy, thereby protecting the heart from progression into heart failure.
Figures
Comment in
-
Atrogin1/MAFbx: what atrophy, hypertrophy, and cardiac failure have in common.Circ Res. 2011 Jul 8;109(2):123-6. doi: 10.1161/CIRCRESAHA.111.248872. Circ Res. 2011. PMID: 21737813 No abstract available.
References
-
- Morisco C, Sadoshima J, Trimarco B, Arora R, Vatner DE, Vatner SF. Is treating cardiac hypertrophy salutary or detrimental: the two faces of Janus. Am J Physiol (Heart Circ Physiol) 2003;284:H1043–H1047. - PubMed
-
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. - PubMed
-
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. - PubMed
-
- Wang X, Robbins J. Heart failure and protein quality control. Circ Res. 2006;99:1315–1328. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HL67724/HL/NHLBI NIH HHS/United States
- R01 HL091469/HL/NHLBI NIH HHS/United States
- P01 AG027211/AG/NIA NIH HHS/United States
- HL69020/HL/NHLBI NIH HHS/United States
- R21 HL098802/HL/NHLBI NIH HHS/United States
- R01 HL067724/HL/NHLBI NIH HHS/United States
- P01 HL059139/HL/NHLBI NIH HHS/United States
- HL102738/HL/NHLBI NIH HHS/United States
- R01 HL112330/HL/NHLBI NIH HHS/United States
- R01 AG023039/AG/NIA NIH HHS/United States
- AG27211/AG/NIA NIH HHS/United States
- R01 HL102738/HL/NHLBI NIH HHS/United States
- HL98802/HL/NHLBI NIH HHS/United States
- HL91469/HL/NHLBI NIH HHS/United States
- P01 HL069020/HL/NHLBI NIH HHS/United States
- HL59139/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
