

Paradynamics: an effective and reliable model for ab initio QM/MM free-energy calculations and related tasks
- PMID: 21618985
- PMCID: PMC3124314
- DOI: 10.1021/jp201217b
Paradynamics: an effective and reliable model for ab initio QM/MM free-energy calculations and related tasks
Abstract
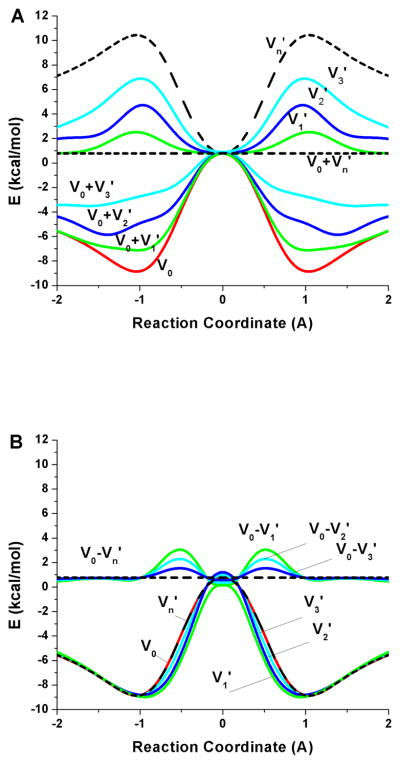
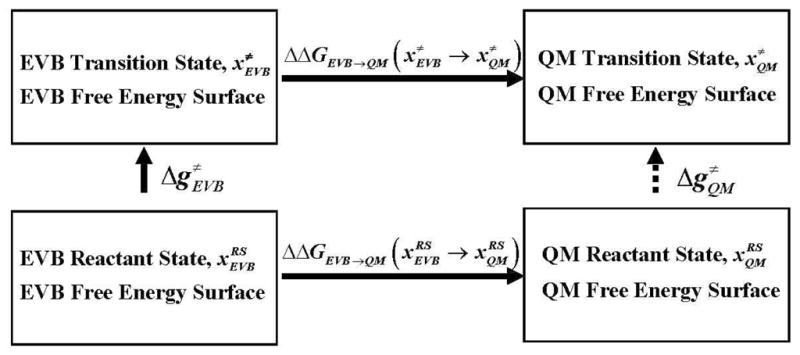
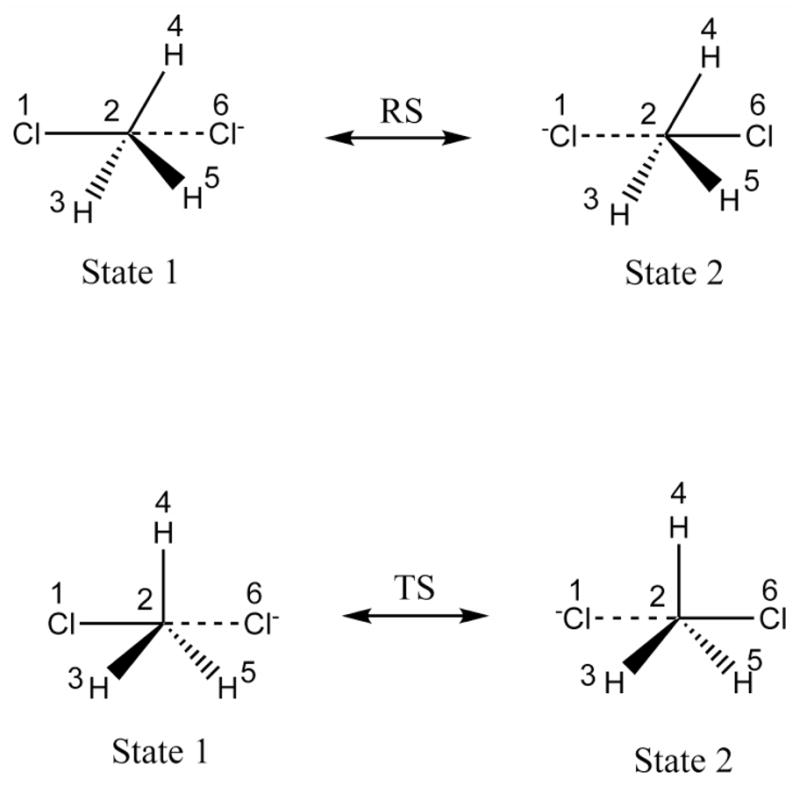
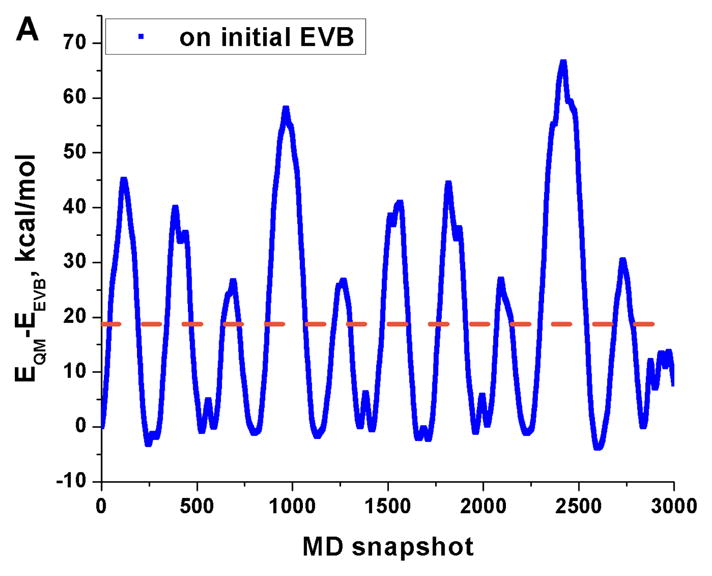
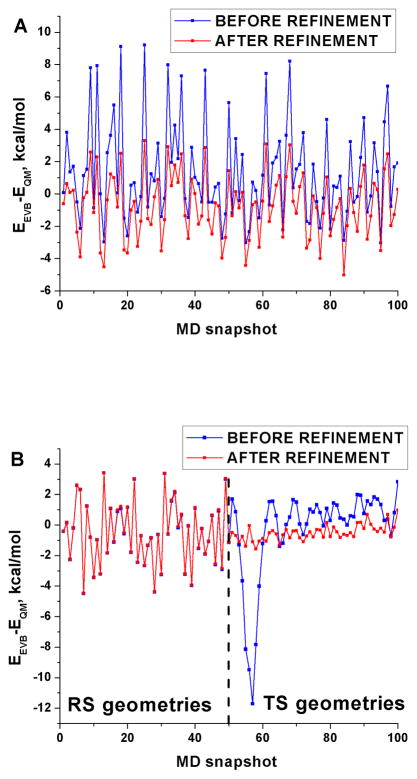
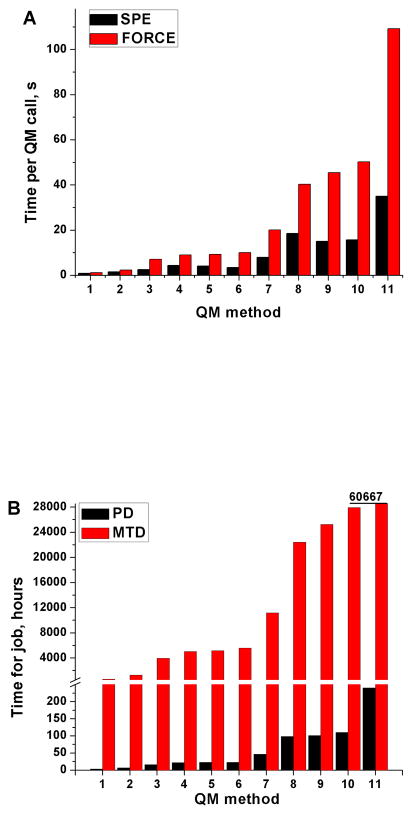
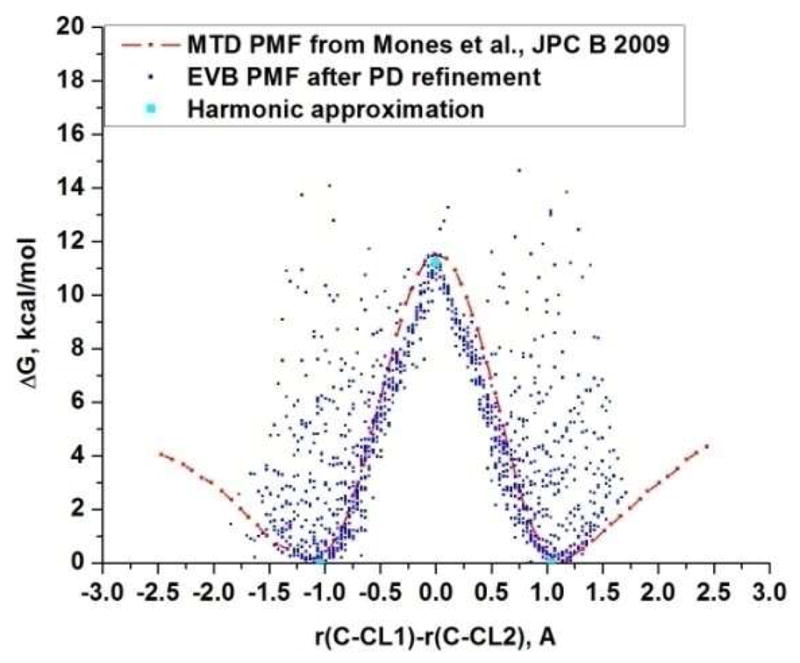
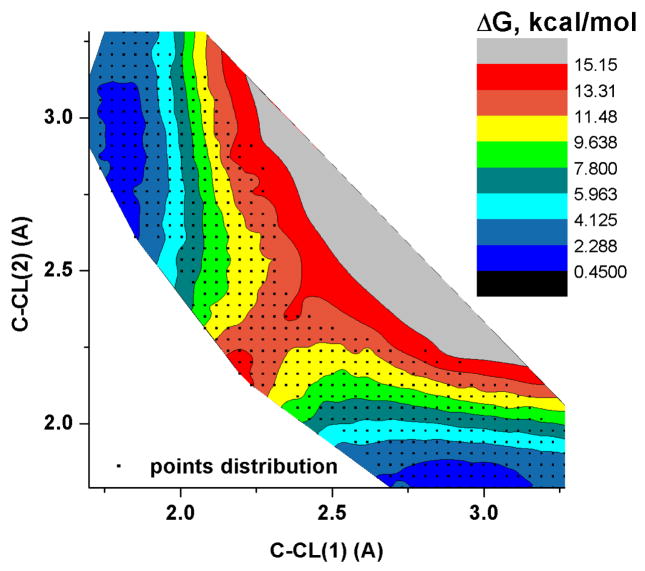
Recent years have seen tremendous effort in the development of approaches with which to obtain quantum mechanics/molecular mechanics (QM/MM) free energies for reactions in the condensed phase. Nevertheless, there remain significant challenges to address, particularly, the high computational cost involved in performing proper configurational sampling and, in particular, in obtaining ab initio QM/MM (QM(ai)/MM) free-energy surfaces. One increasingly popular approach that seems to offer an ideal way to progress in this direction is the elegant metadynamics (MTD) approach. However, in the current work, we point out the subtle efficiency problems associated with this approach and illustrate that we have at hand what is arguably a more powerful approach. More specifically, we demonstrate the effectiveness of an updated version of our original idea of using a classical reference potential for QM(ai)/MM calculations [J. Phys. Chem. 1995, 99, 17516)], which we refer to as paradynamics (PD). This approach is based on the use of an empirical valence bond (EVB) reference potential, which is already similar to the real ab initio potential. The reference potential is fitted to the ab initio potential by an iterative and, to a great degree, automated refinement procedure. The corresponding free-energy profile is then constructed using the refined EVB potential, and the linear response approximation (LRA) is used to evaluate the QM(ai)/MM activation free-energy barrier. The automated refinement of the EVB surface (and thus the reduction of the difference between the reference and ab initio potentials) is a key factor in accelerating the convergence of the LRA approach. We apply our PD approach to a test reaction, namely, the S(N)2 reaction between a chloride ion and methyl chloride, and demonstrate that, at present, this approach is far more powerful and cost-effective than the metadynamics approach (at least in its current implementation). We also discuss the general features of the PD approach in terms of its ability to explore complex systems and clarify that it is not a specialized approach limited to only accelerating QM(ai)/MM calculations with proper sampling, but rather can be used in a wide variety of applications. In fact, we point out that the use of a reference (CG) potential coupled with its PD refinement, as well as our renormalization approach, provides very general and powerful strategies that can be used very effectively to explore any property that has been studied by the MTD approach.
Figures





References
-
- Bentzien J, Muller RP, Florian J, Warshel A. J Phys Chem B. 1998;102:2293–2301.
-
- Štrajbl M, Hong G, Warshel A. J Phys Chem B. 2002;106:13333–13343.
-
- Rosta E, Klähn M, Warshel A. J Phys Chem B. 2006;110:2934–2941. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
