

Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo
- PMID: 21619641
- PMCID: PMC3224557
- DOI: 10.1186/1750-1326-6-36
Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo
Abstract
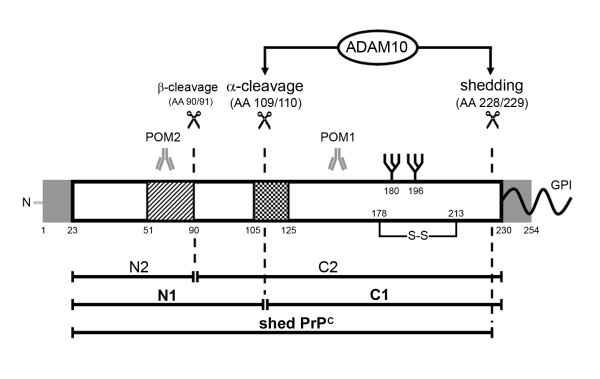
Background: The cellular prion protein (PrPC) fulfils several yet not completely understood physiological functions. Apart from these functions, it has the ability to misfold into a pathogenic scrapie form (PrPSc) leading to fatal transmissible spongiform encephalopathies. Proteolytic processing of PrPC generates N- and C-terminal fragments which play crucial roles both in the pathophysiology of prion diseases and in transducing physiological functions of PrPC. A-disintegrin-and-metalloproteinase 10 (ADAM10) has been proposed by cell culture experiments to be responsible for both shedding of PrPC and its α-cleavage. Here, we analyzed the role of ADAM10 in the proteolytic processing of PrPC in vivo.
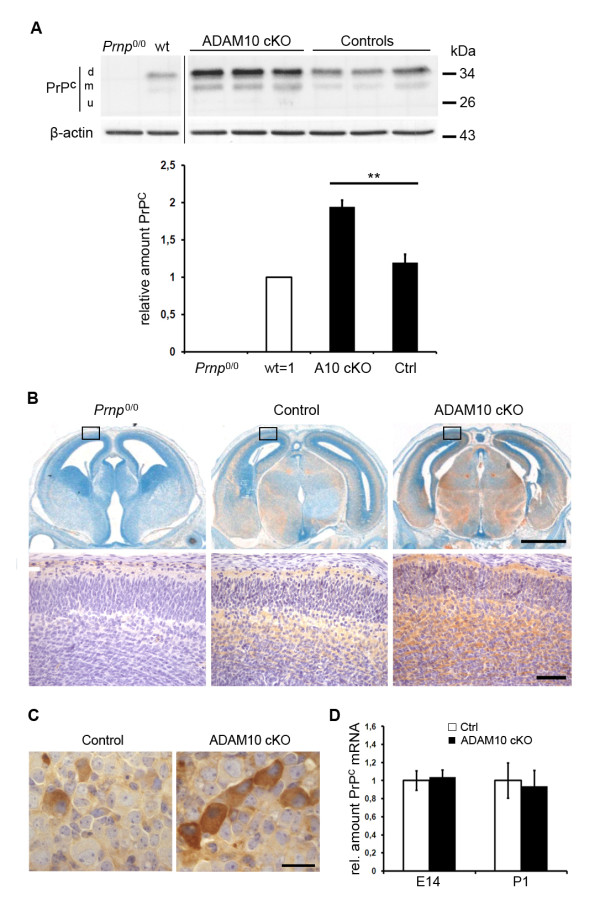
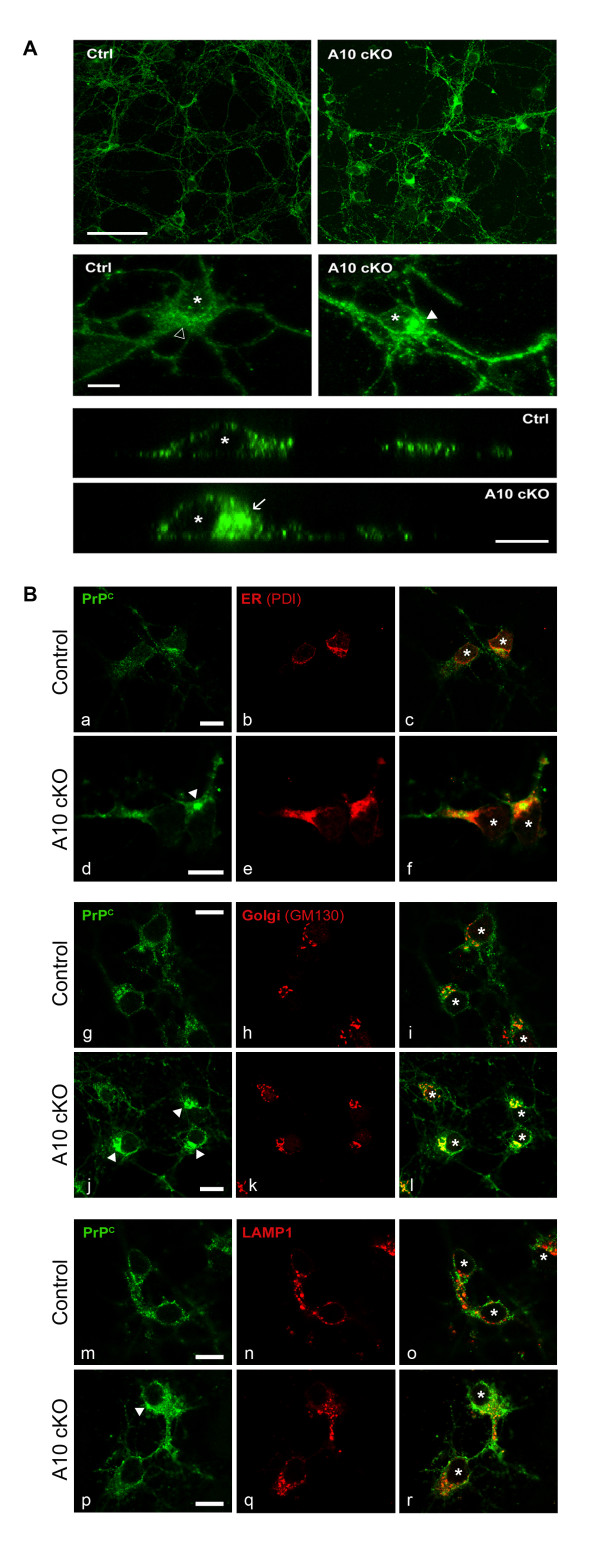
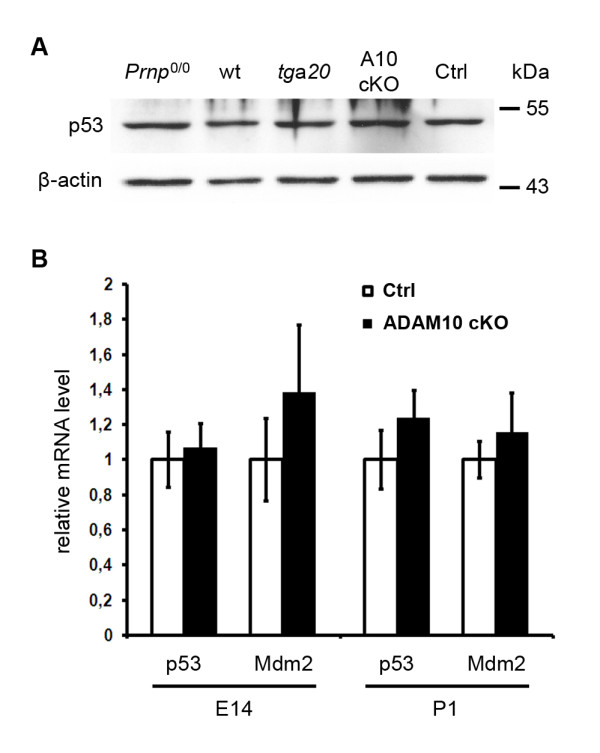
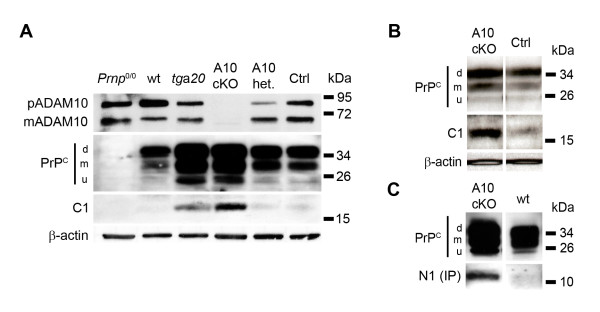
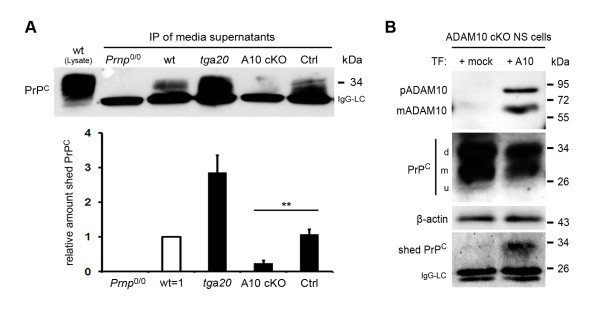
Results: Using neuron-specific Adam10 knockout mice, we show that ADAM10 is the sheddase of PrPC and that its absence in vivo leads to increased amounts and accumulation of PrPC in the early secretory pathway by affecting its posttranslational processing. Elevated PrPC levels do not induce apoptotic signalling via p53. Furthermore, we show that ADAM10 is not responsible for the α-cleavage of PrPC.
Conclusion: Our study elucidates the proteolytic processing of PrPC and proves a role of ADAM10 in shedding of PrPC in vivo. We suggest that ADAM10 is a mediator of PrPC homeostasis at the plasma membrane and, thus, might be a regulator of the multiple functions discussed for PrPC. Furthermore, identification of ADAM10 as the sheddase of PrPC opens the avenue to devising novel approaches for therapeutic interventions against prion diseases.
Figures
References
-
- Sunyach C, Cisse MA, da Costa CA, Vincent B, Checler F. The C-terminal products of cellular prion protein processing, C1 and C2, exert distinct influence on p53-dependent staurosporine-induced caspase-3 activation. J Biol Chem. 2007;282(3):1956–1963. - PubMed
-
- Chesebro B, Race B, Meade-White K, Lacasse R, Race R, Klingeborn M, Striebel J, Dorward D, McGovern G, Jeffrey M. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog. 2010;6(3):e1000800. doi: 10.1371/journal.ppat.1000800. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
