

DASH: a method for identical-by-descent haplotype mapping uncovers association with recent variation
- PMID: 21620352
- PMCID: PMC3113343
- DOI: 10.1016/j.ajhg.2011.04.023
DASH: a method for identical-by-descent haplotype mapping uncovers association with recent variation
Abstract
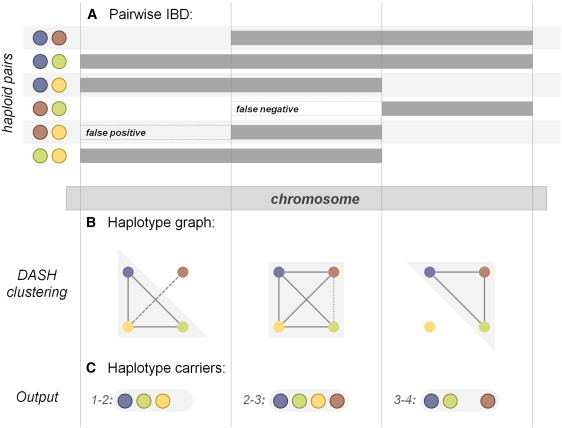
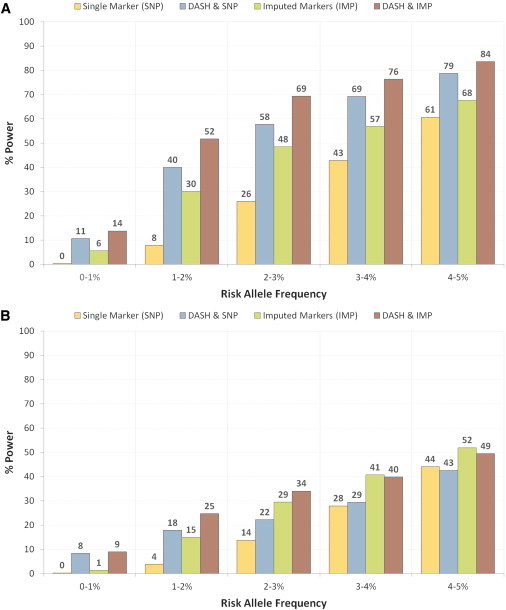
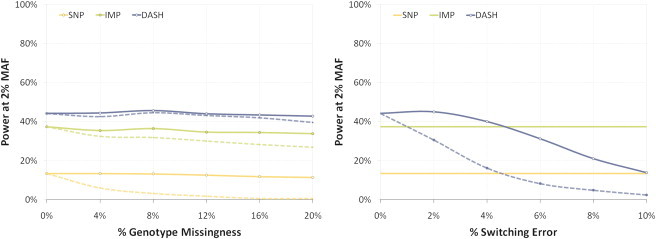
Rare variants affecting phenotype pose a unique challenge for human genetics. Although genome-wide association studies have successfully detected many common causal variants, they are underpowered in identifying disease variants that are too rare or population-specific to be imputed from a general reference panel and thus are poorly represented on commercial SNP arrays. We set out to overcome these challenges and detect association between disease and rare alleles using SNP arrays by relying on long stretches of genomic sharing that are identical by descent. We have developed an algorithm, DASH, which builds upon pairwise identical-by-descent shared segments to infer clusters of individuals likely to be sharing a single haplotype. DASH constructs a graph with nodes representing individuals and links on the basis of such segments spanning a locus and uses an iterative minimum cut algorithm to identify densely connected components. We have applied DASH to simulated data and diverse GWAS data sets by constructing haplotype clusters and testing them for association. In simulations we show this approach to be significantly more powerful than single-marker testing in an isolated population that is from Kosrae, Federated States of Micronesia and has abundant IBD, and we provide orthogonal information for rare, recent variants in the outbred Wellcome Trust Case-Control Consortium (WTCCC) data. In both cohorts, we identified a number of haplotype associations, five such loci in the WTCCC data and ten in the isolated, that were conditionally significant beyond any individual nearby markers. We have replicated one of these loci in an independent European cohort and identified putative structural changes in low-pass whole-genome sequence of the cluster carriers.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Browning B.L., Browning S.R. Efficient multilocus association testing for whole genome association studies using localized haplotype clustering. Genet. Epidemiol. 2007;31:365–375. - PubMed
-
- Zaykin D.V., Westfall P.H., Young S.S., Karnoub M.A., Wagner M.J., Ehm M.G. Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum. Hered. 2002;53:79–91. - PubMed
-
- Purcell S., Daly M.J., Sham P.C. WHAP: Haplotype-based association analysis. Bioinformatics. 2007;23:255–256. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
