

Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy
- PMID: 21620606
- PMCID: PMC3742010
- DOI: 10.1016/j.jacc.2010.12.044
Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy
Abstract
Objectives: We investigated the effect of reducing mitochondrial oxidative stress by the mitochondrial-targeted antioxidant peptide SS-31 in hypertensive cardiomyopathy.
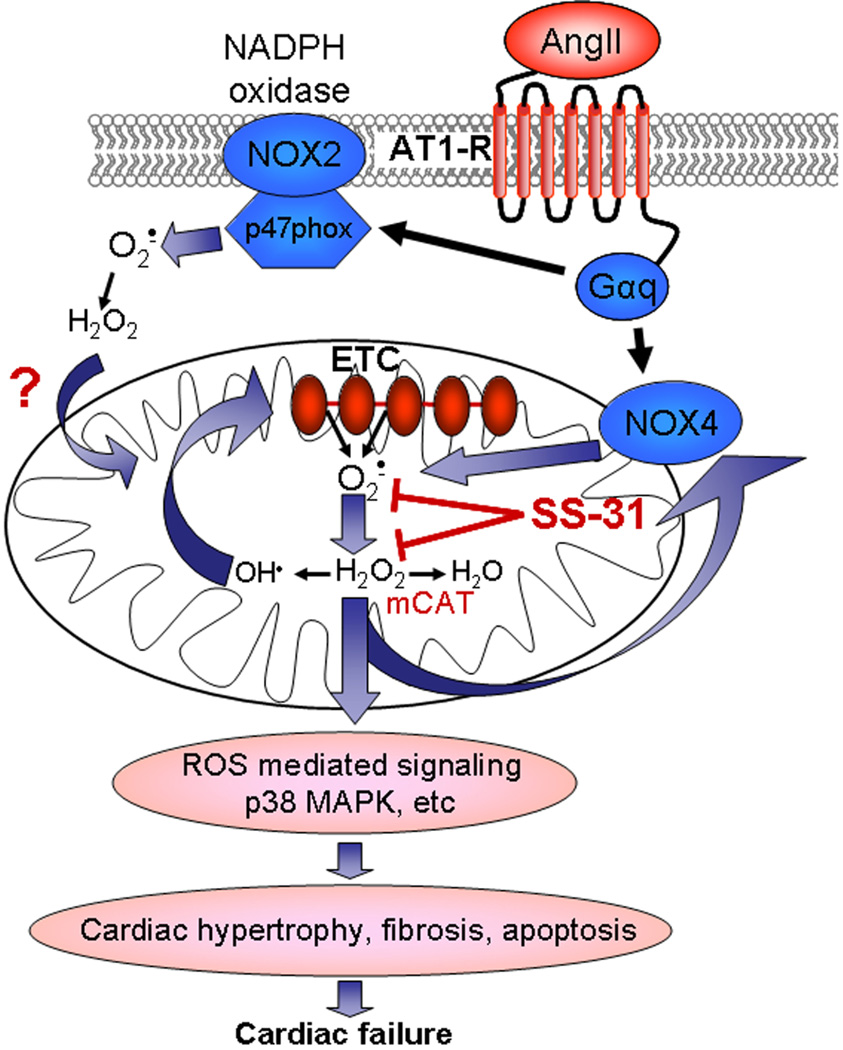
Background: Oxidative stress has been implicated in hypertensive cardiovascular diseases. Mitochondria and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase have been proposed as primary sites of reactive oxygen species (ROS) generation.
Methods: The mitochondrial targeted antioxidant peptide SS-31 was used to determine the role of mitochondrial oxidative stress in angiotensin II (Ang)-induced cardiomyopathy as well as in Gαq overexpressing mice with heart failure.
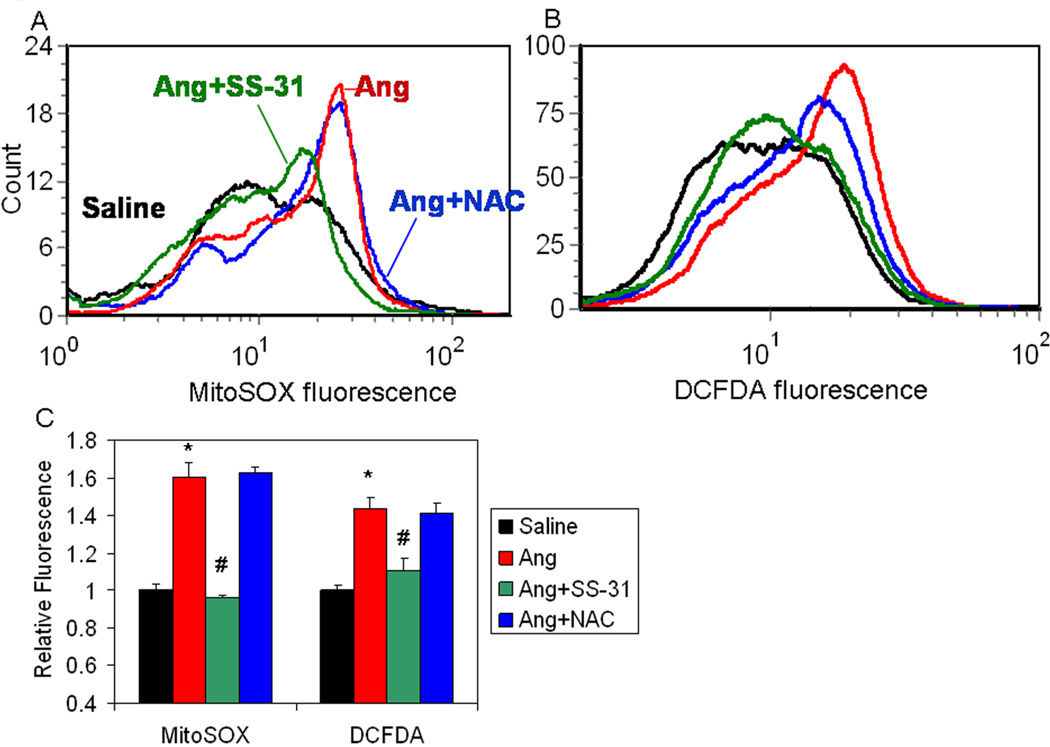
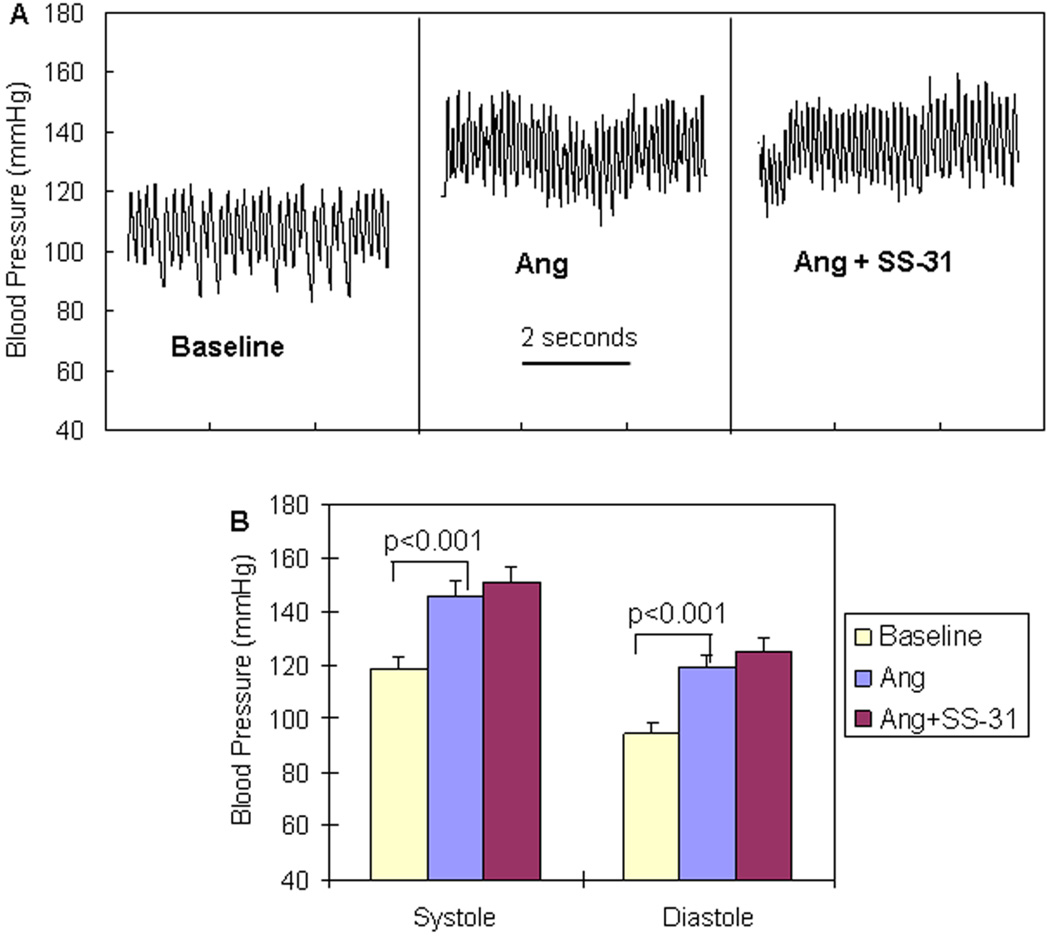
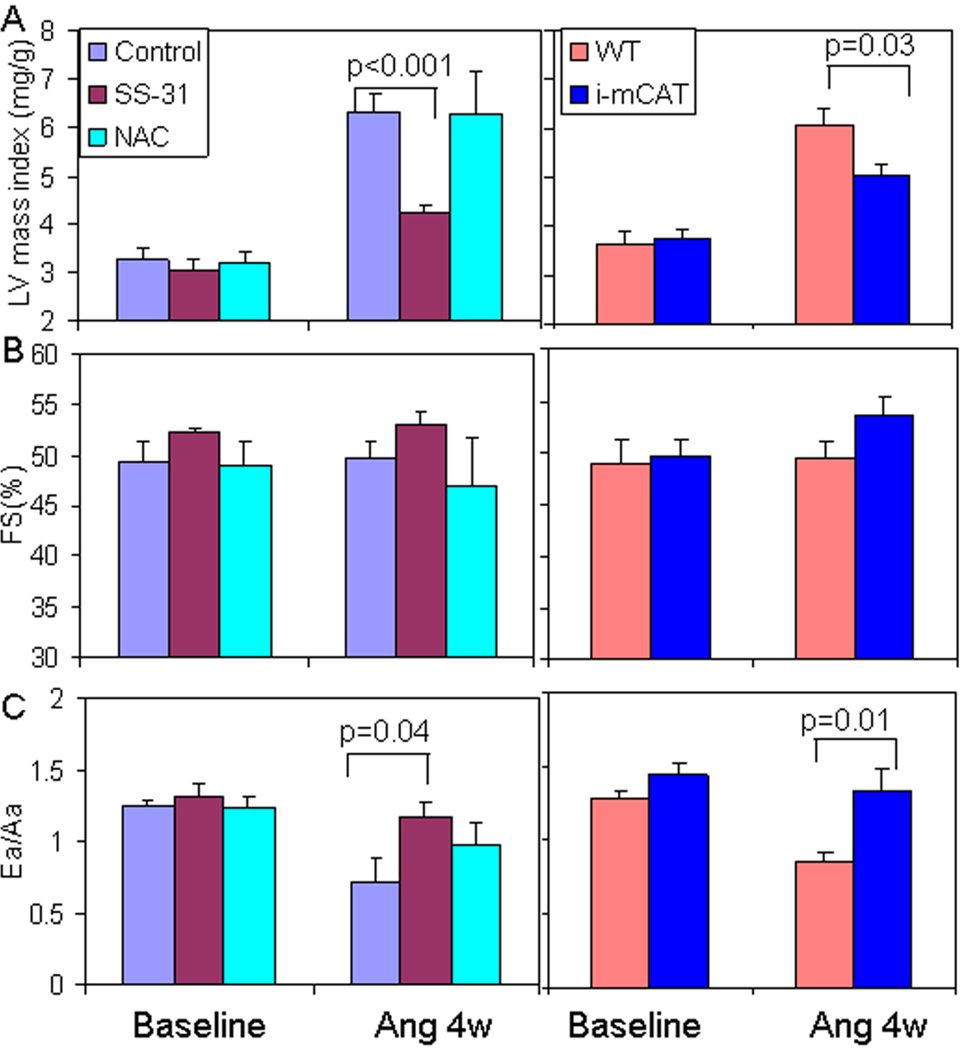
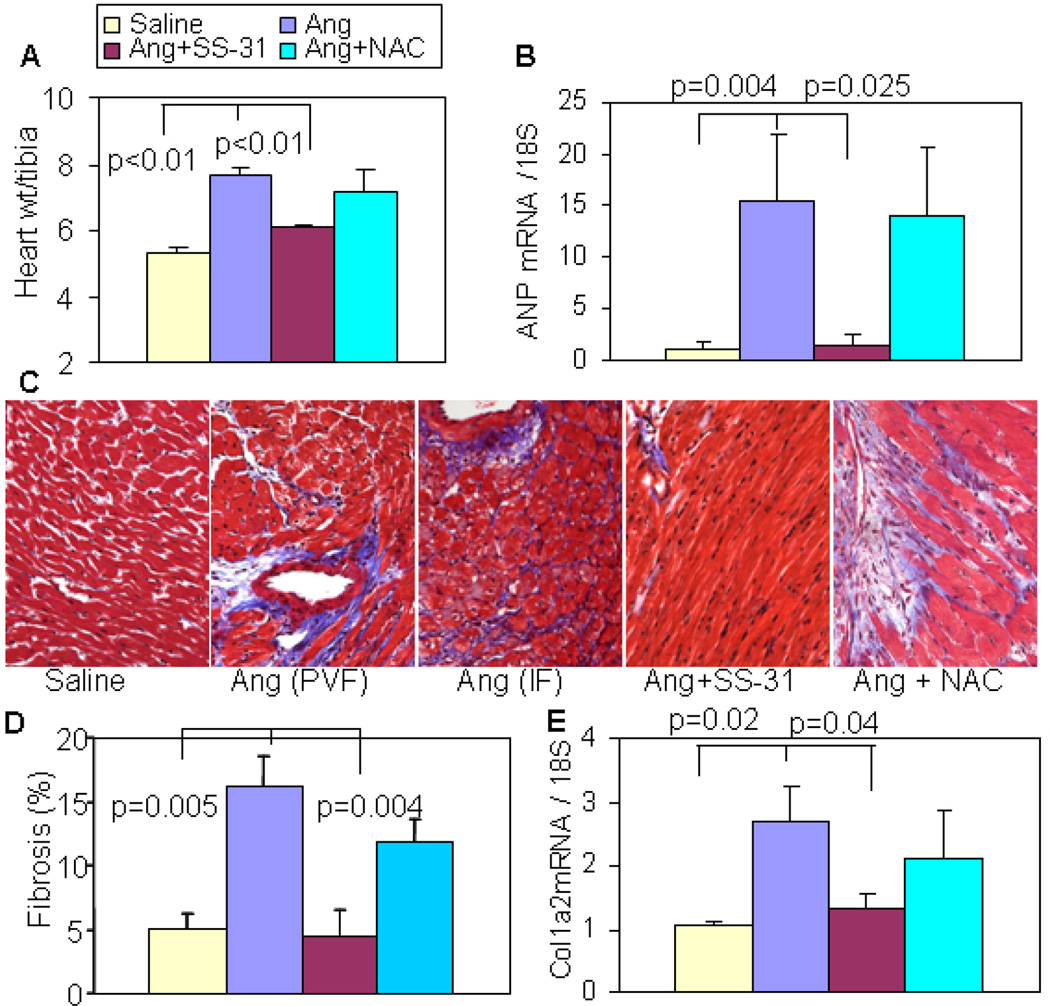
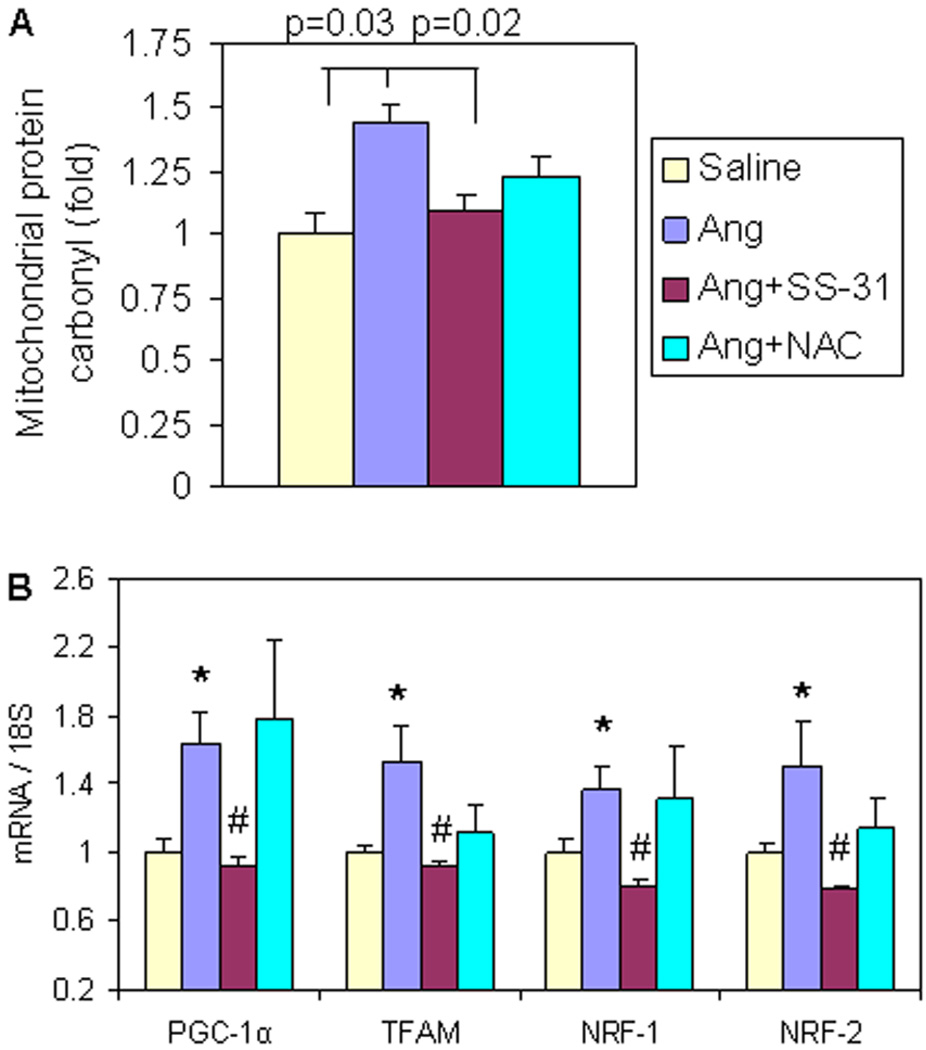
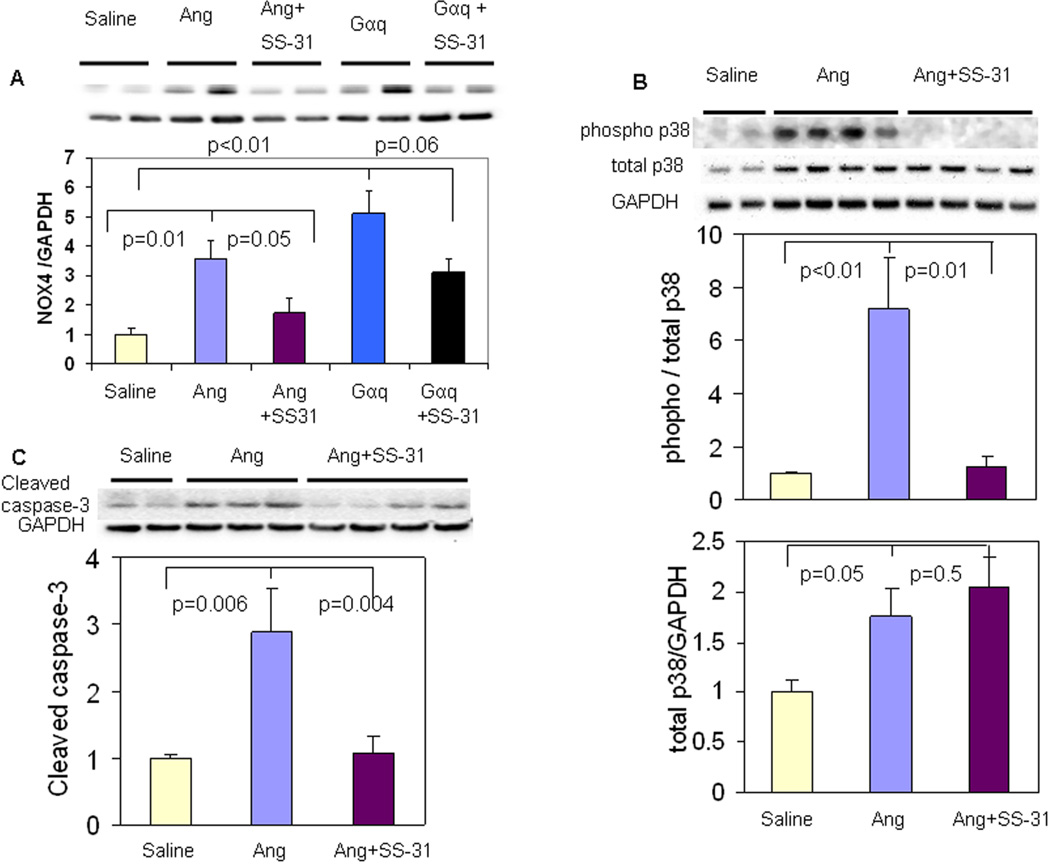
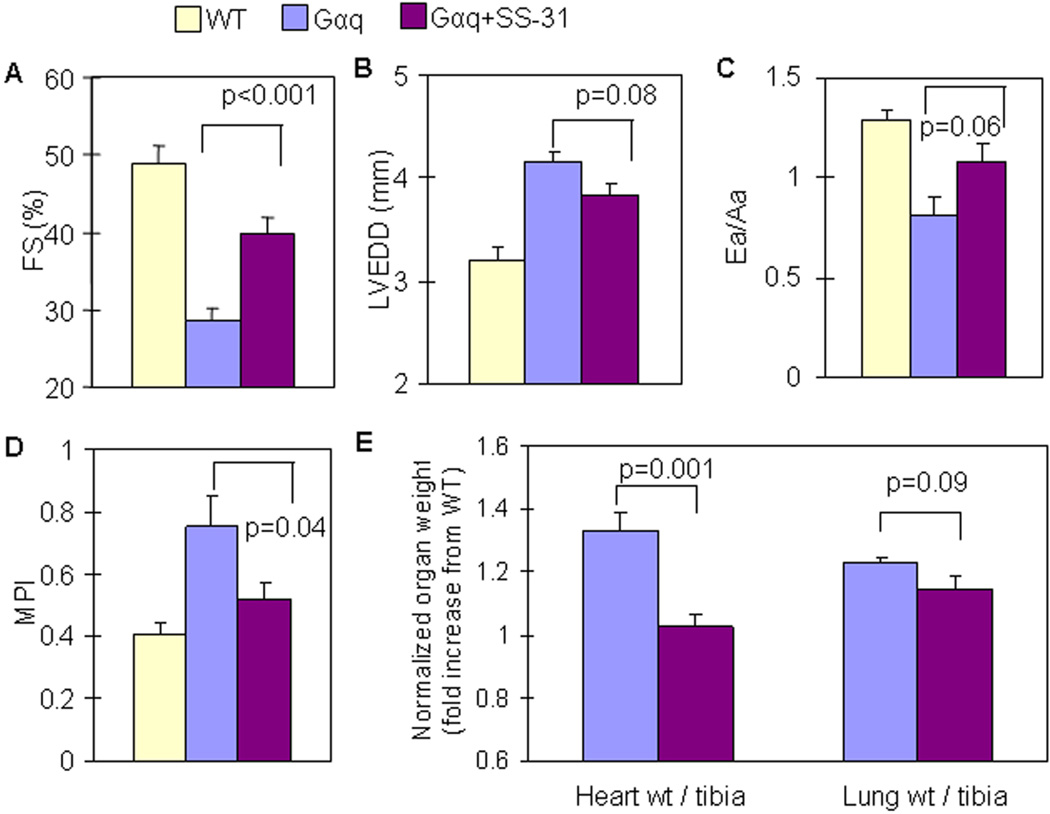
Results: Ang induces mitochondrial ROS in neonatal cardiomyocytes, which is prevented by SS-31, but not the nontargeted antioxidant N-acetyl cysteine (NAC). Continuous administration of Ang for 4 weeks in mice significantly increased both systolic and diastolic blood pressure, and this was not affected by SS-31 treatment. Ang was associated with up-regulation of NADPH oxidase 4 (NOX4) expression and increased cardiac mitochondrial protein oxidative damage, and induced the signaling for mitochondrial biogenesis. Reducing mitochondrial ROS by SS-31 substantially attenuated Ang-induced NOX4 up-regulation, mitochondrial oxidative damage, up-regulation of mitochondrial biogenesis, and phosphorylation of p38 mitogen-activated protein kinase and prevented apoptosis, concomitant with amelioration of Ang-induced cardiac hypertrophy, diastolic dysfunction, and fibrosis, despite the absence of blood pressure-lowering effect. The NAC did not show any beneficial effect. The SS-31 administration for 4 weeks also partially rescued the heart failure phenotype of Gαq overexpressing mice.
Conclusions: Mitochondrial targeted peptide SS-31 ameliorates cardiomyopathy resulting from prolonged Ang stimulation as well as Gαq overexpression, suggesting its potential clinical application for target organ protection in hypertensive cardiovascular diseases.
Copyright © 2011 American College of Cardiology Foundation. Published by Elsevier Inc. All rights reserved.
Figures
Comment in
-
Targeting mitochondrial oxidative stress in heart failure throttling the afterburner.J Am Coll Cardiol. 2011 Jun 28;58(1):83-6. doi: 10.1016/j.jacc.2011.01.032. Epub 2011 May 27. J Am Coll Cardiol. 2011. PMID: 21620605 No abstract available.
References
-
- Lawes CM, Vander Hoorn S, Rodgers A. Global burden of blood-pressure-related disease, 2001. Lancet. 2008;371:1513–1518. - PubMed
-
- Gardin JM, Lauer MS. Left ventricular hypertrophy: the next treatable, silent killer? Jama. 2004;292:2396–2398. - PubMed
-
- Mollnau H, Wendt M, Szocs K, et al. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res. 2002;90:E58–E65. - PubMed
-
- Kimura S, Zhang GX, Nishiyama A, et al. Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension. 2005;45:438–444. - PubMed
-
- Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin IImediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
