

Protective role of heme oxygenase-1 against inflammation in atherosclerosis
- PMID: 21622183
- PMCID: PMC5940339
- DOI: 10.2741/3860
Protective role of heme oxygenase-1 against inflammation in atherosclerosis
Abstract
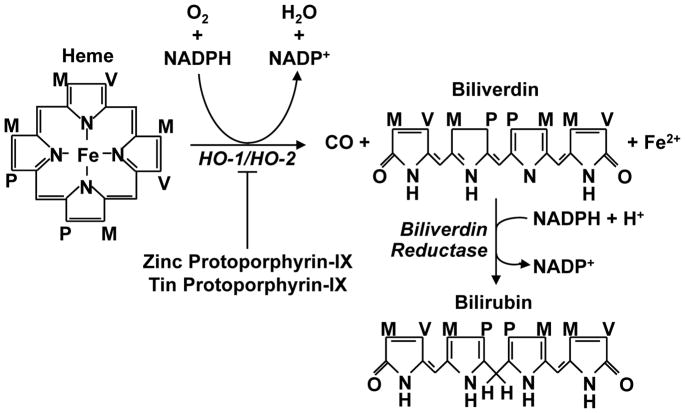
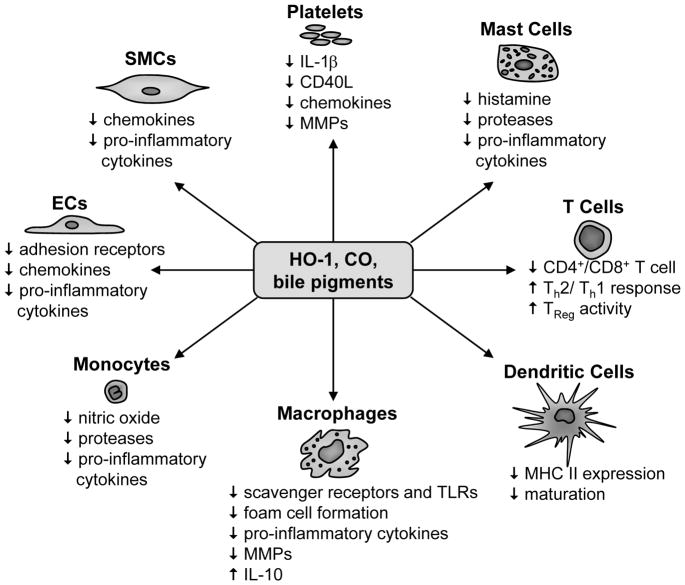
Heme oxygenase-1 (HO-1) catalyzes the first and rate-limiting step in the metabolism of free heme into equimolar amounts of ferrous iron, carbon monoxide (CO), and biliverdin. Biliverdin is subsequently converted to bilirubin by biliverdin reductase. HO-1 has recently been identified as a promising therapeutic target in the treatment of vascular inflammatory disease, including atherosclerosis. HO-1 represses inflammation by removing the pro-inflammatory molecule heme and by generating CO and the bile pigments, biliverdin and bilirubin. These HO-1 reaction products are capable of blocking innate and adaptive immune responses by modifying the activation, differentiation, maturation, and/or polarization of numerous immune cells, including endothelial cells, monocytes/macrophages, dendritic cells, T lymphocytes, mast cells, and platelets. These cellular actions by CO and bile pigments result in diminished leukocyte recruitment and infiltration, and pro-inflammatory mediator production within atherosclerotic lesions. This review highlights the mechanisms by which HO-1 suppresses vascular inflammation in atherosclerosis, and explores possible therapeutic modalities by which HO-1 and its reaction products can be employed to ameliorate vascular inflammatory disease.
Figures
References
-
- Faxon DP, Fuster V, Libby P, Beckman JA, Hiatt WR, Thompson RW, Topper JN, Annex BH, Rundback JH, Fabunmi RP, Robertson RM, Loscalzo J. American Heart Association: Atherosclerotic Vascular Disease Conference: Writing Group III: pathophysiology. Circulation. 2004;109:2617–2625. - PubMed
-
- Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, Hailpern SM, Ho MM, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O’Donnell C, Roger V, Sorlie P, Steinberger J, Thom T, Wilson M, Hong Y. American Heart Association: Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25–e146. - PubMed
-
- Olshansky SJ, Passaro DJ, Hershow RC, Layden J, Carnes BA, Brody J, Hayflick L, Butler RN, Allison DB, Ludwig DS. A potential decline in life expectancy in the United States in the 21st century. N Engl J Med. 2005;352:1138–1145. - PubMed
-
- Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–519. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
