

An integrated approach to characterize genetic interaction networks in yeast metabolism
- PMID: 21623372
- PMCID: PMC3125439
- DOI: 10.1038/ng.846
An integrated approach to characterize genetic interaction networks in yeast metabolism
Abstract
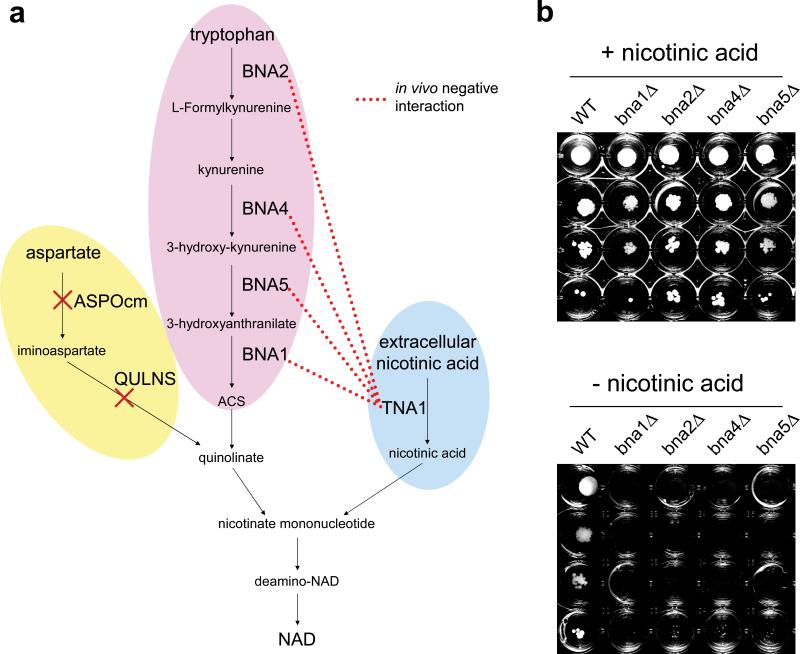
Although experimental and theoretical efforts have been applied to globally map genetic interactions, we still do not understand how gene-gene interactions arise from the operation of biomolecular networks. To bridge the gap between empirical and computational studies, we i, quantitatively measured genetic interactions between ∼185,000 metabolic gene pairs in Saccharomyces cerevisiae, ii, superposed the data on a detailed systems biology model of metabolism and iii, introduced a machine-learning method to reconcile empirical interaction data with model predictions. We systematically investigated the relative impacts of functional modularity and metabolic flux coupling on the distribution of negative and positive genetic interactions. We also provide a mechanistic explanation for the link between the degree of genetic interaction, pleiotropy and gene dispensability. Last, we show the feasibility of automated metabolic model refinement by correcting misannotations in NAD biosynthesis and confirming them by in vivo experiments.
Figures
References
-
- Schuldiner M, et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 2005;123:507–19. - PubMed
-
- Collins SR, et al. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature. 2007;446:806–10. - PubMed
-
- Tong AH, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–13. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HG005084/HG/NHGRI NIH HHS/United States
- 084314/WT_/Wellcome Trust/United Kingdom
- BB/C505140/2/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/C505140/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 1R01HG005853-01/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
