

deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data
- PMID: 21625565
- PMCID: PMC3098195
- DOI: 10.1371/journal.pcbi.1001138
deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data
Abstract
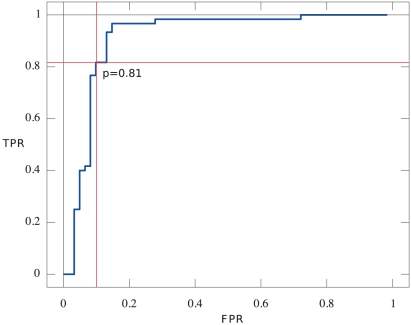
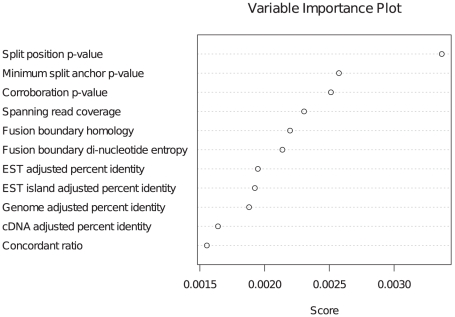
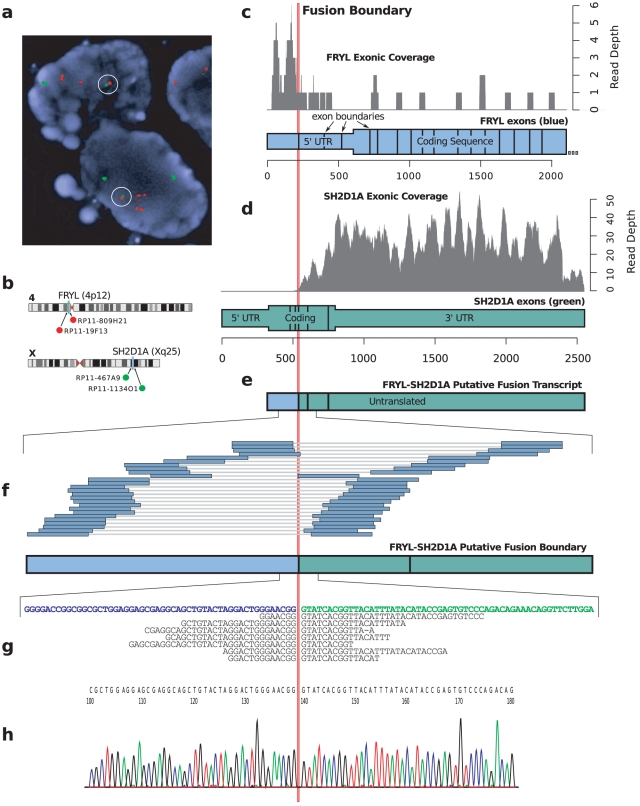
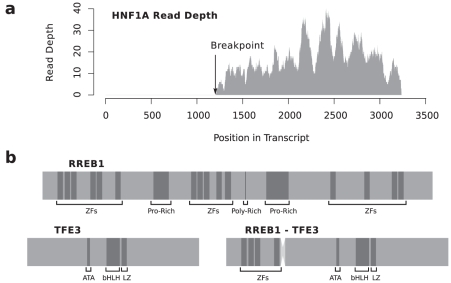
Gene fusions created by somatic genomic rearrangements are known to play an important role in the onset and development of some cancers, such as lymphomas and sarcomas. RNA-Seq (whole transcriptome shotgun sequencing) is proving to be a useful tool for the discovery of novel gene fusions in cancer transcriptomes. However, algorithmic methods for the discovery of gene fusions using RNA-Seq data remain underdeveloped. We have developed deFuse, a novel computational method for fusion discovery in tumor RNA-Seq data. Unlike existing methods that use only unique best-hit alignments and consider only fusion boundaries at the ends of known exons, deFuse considers all alignments and all possible locations for fusion boundaries. As a result, deFuse is able to identify fusion sequences with demonstrably better sensitivity than previous approaches. To increase the specificity of our approach, we curated a list of 60 true positive and 61 true negative fusion sequences (as confirmed by RT-PCR), and have trained an adaboost classifier on 11 novel features of the sequence data. The resulting classifier has an estimated value of 0.91 for the area under the ROC curve. We have used deFuse to discover gene fusions in 40 ovarian tumor samples, one ovarian cancer cell line, and three sarcoma samples. We report herein the first gene fusions discovered in ovarian cancer. We conclude that gene fusions are not infrequent events in ovarian cancer and that these events have the potential to substantially alter the expression patterns of the genes involved; gene fusions should therefore be considered in efforts to comprehensively characterize the mutational profiles of ovarian cancer transcriptomes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
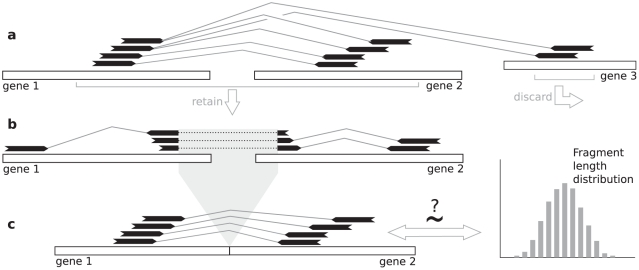
 . e) The similar fragment length condition is the constraint that
. e) The similar fragment length condition is the constraint that  must be no more than
must be no more than  .
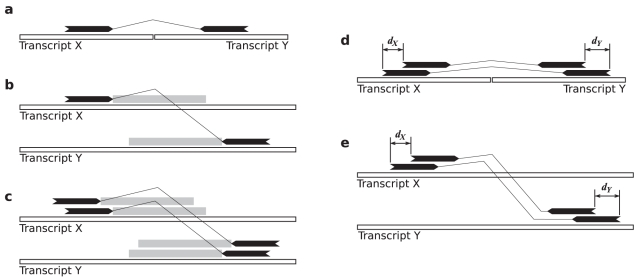
.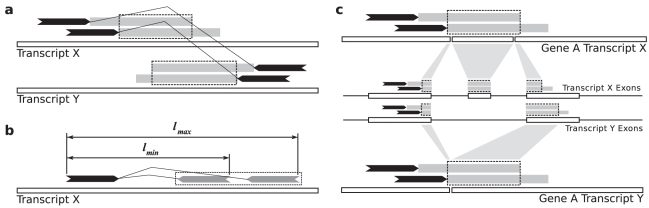
 is projected into transcript
is projected into transcript  by remapping the start of the approximate fusion boundary from
by remapping the start of the approximate fusion boundary from  , to the genome, to
, to the genome, to  .
.
References
-
- Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–599. - PubMed
-
- Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233–245. - PubMed
-
- Tognon C, Knezevich SR, Huntsman D, Roskelley CD, Melnyk N, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2:367–376. - PubMed
-
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
