

Faster Smith-Waterman database searches with inter-sequence SIMD parallelisation
- PMID: 21631914
- PMCID: PMC3120707
- DOI: 10.1186/1471-2105-12-221
Faster Smith-Waterman database searches with inter-sequence SIMD parallelisation
Abstract
Background: The Smith-Waterman algorithm for local sequence alignment is more sensitive than heuristic methods for database searching, but also more time-consuming. The fastest approach to parallelisation with SIMD technology has previously been described by Farrar in 2007. The aim of this study was to explore whether further speed could be gained by other approaches to parallelisation.
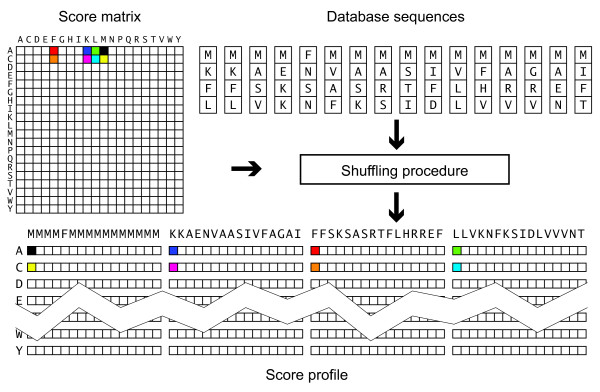
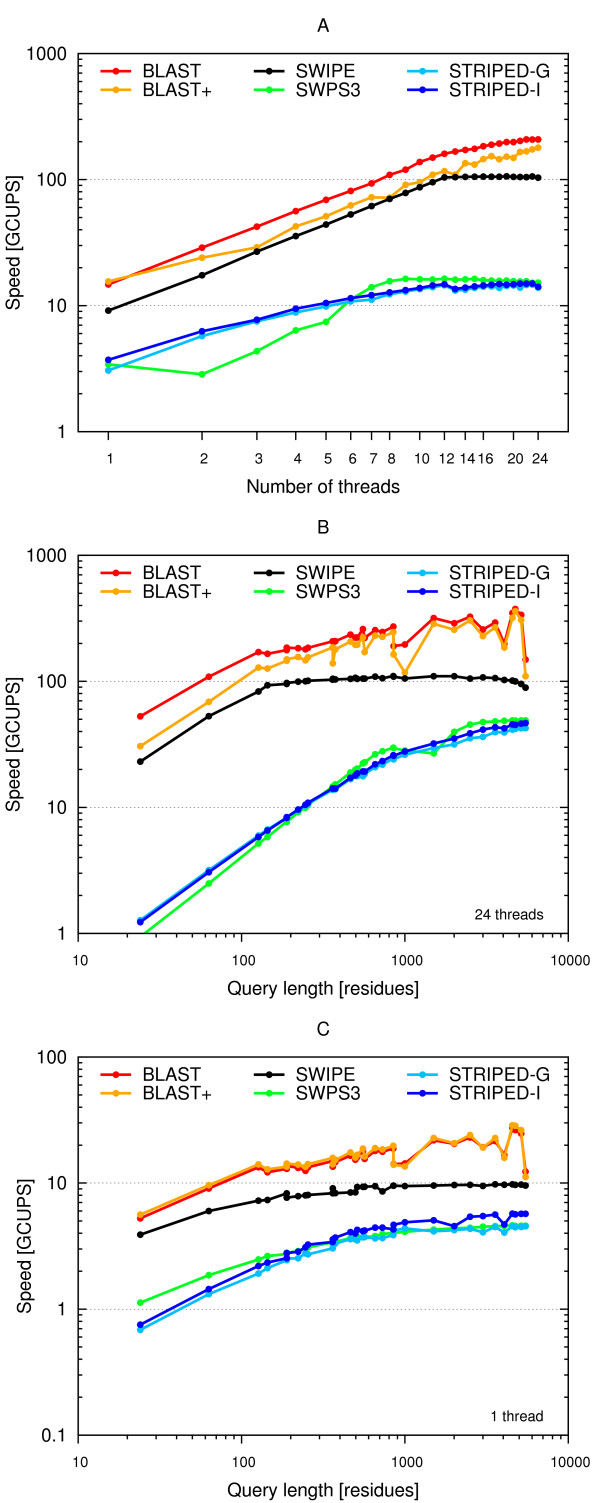
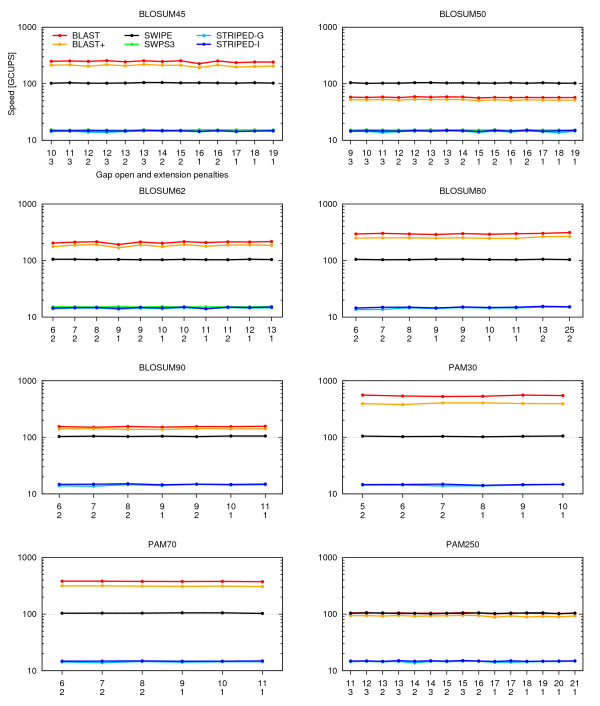
Results: A faster approach and implementation is described and benchmarked. In the new tool SWIPE, residues from sixteen different database sequences are compared in parallel to one query residue. Using a 375 residue query sequence a speed of 106 billion cell updates per second (GCUPS) was achieved on a dual Intel Xeon X5650 six-core processor system, which is over six times more rapid than software based on Farrar's 'striped' approach. SWIPE was about 2.5 times faster when the programs used only a single thread. For shorter queries, the increase in speed was larger. SWIPE was about twice as fast as BLAST when using the BLOSUM50 score matrix, while BLAST was about twice as fast as SWIPE for the BLOSUM62 matrix. The software is designed for 64 bit Linux on processors with SSSE3. Source code is available from http://dna.uio.no/swipe/ under the GNU Affero General Public License.
Conclusions: Efficient parallelisation using SIMD on standard hardware makes it possible to run Smith-Waterman database searches more than six times faster than before. The approach described here could significantly widen the potential application of Smith-Waterman searches. Other applications that require optimal local alignment scores could also benefit from improved performance.
Figures
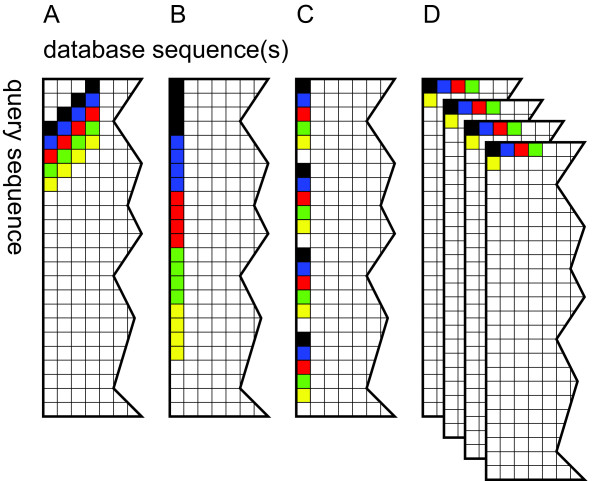
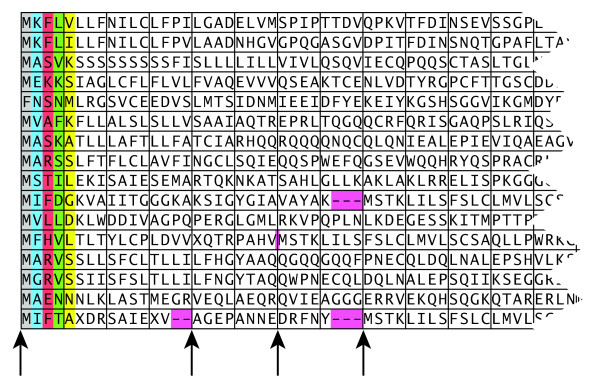
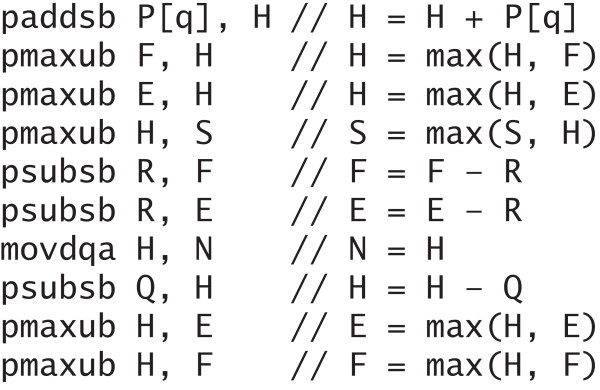
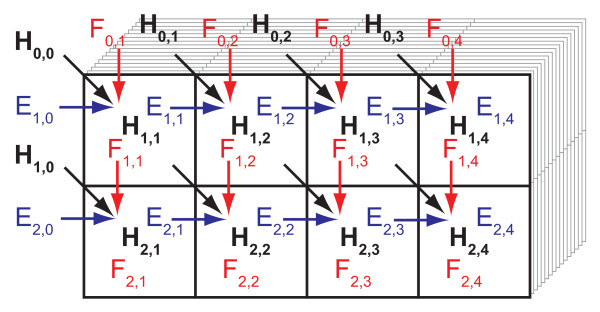
References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
