

Survival and associated factors in 268 adults with Pompe disease prior to treatment with enzyme replacement therapy
- PMID: 21631931
- PMCID: PMC3135500
- DOI: 10.1186/1750-1172-6-34
Survival and associated factors in 268 adults with Pompe disease prior to treatment with enzyme replacement therapy
Abstract
Background: Pompe disease is a rare lysosomal storage disorder characterized by muscle weakness and wasting. The majority of adult patients have slowly progressive disease, which gradually impairs mobility and respiratory function and may lead to wheelchair and ventilator dependency. It is as yet unknown to what extent the disease reduces the life span of these patients. Our objective was to determine the survival of adults with Pompe disease not receiving ERT and to identify prognostic factors associated with survival.
Methods: Data of 268 patients were collected in a prospective international observational study conducted between 2002 and 2009. Survival analyses from time of diagnosis and from time of study entry were performed using Kaplan-Meier curves and Cox-proportional-hazards regression.
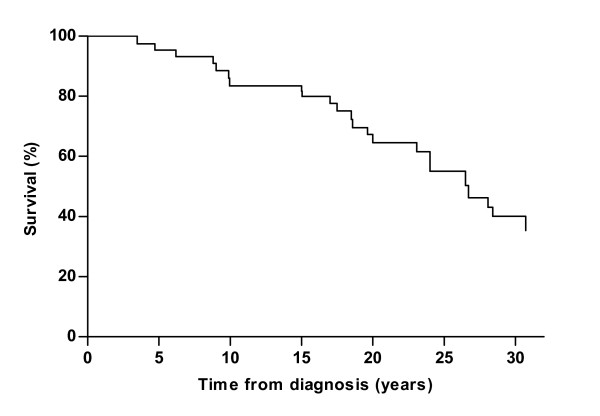
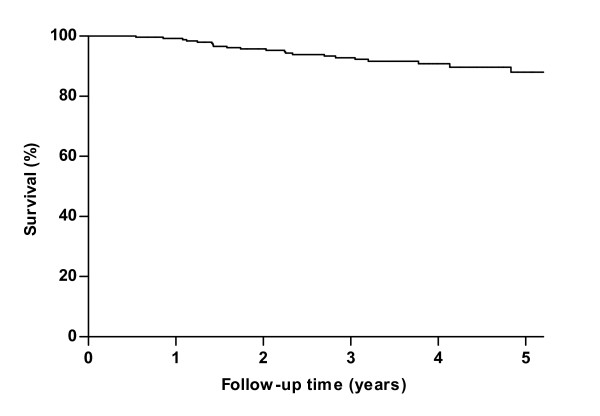
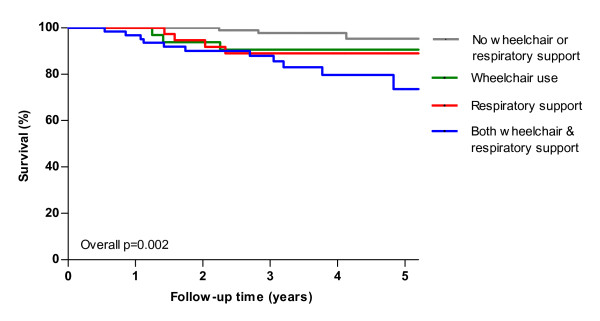
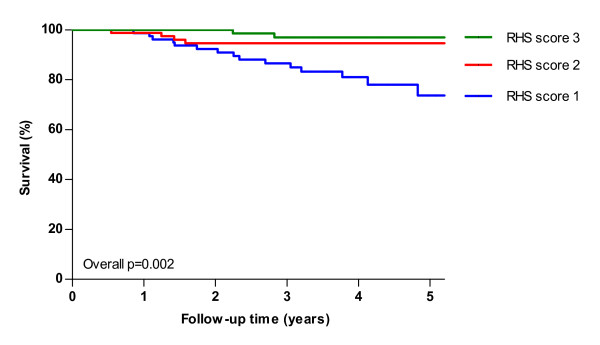
Results: Median age at study entry was 48 years (range 19-79 years). Median survival after diagnosis was 27 years, while median age at diagnosis was 38 years. During follow-up, twenty-three patients died prior to ERT, with a median age at death of 55 (range 23-77 years). Use of wheelchair and/or respiratory support and patients' score on the Rotterdam Handicap Scale (RHS) were identified as prognostic factors for survival. Five-year survival for patients without a wheelchair or respiratory support was 95% compared to 74% in patients who were wheelchair-bound and used respiratory support. In a Dutch subgroup of 99 patients, we compared the observed number of deaths to the expected number of deaths in the age- and sex-matched general population. During a median follow-up of 2.3 years, the number of deaths among the Dutch Pompe patients was higher than the expected number of deaths in the general population.
Conclusion: Our study shows for the first time that untreated adults with Pompe disease have a higher mortality than the general population and that their levels of disability and handicap/participation are the most important factors associated with mortality. These results may be of relevance when addressing the effect of ERT or other potential treatment options on survival.
Figures
References
-
- Hirschhorn R, Reuser AJ. In: The Metabolic and Molecular Bases of Inherited Disease. 8. CR Scriver BA, Sly W, Valle D, editor. New York McGraw-Hill; 2001. Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency; pp. 3389–3420.
-
- Engel A, Hirschhorn R, Huie ML. In: Myology. 3. Engel A F-AC, editor. New York: McGraw-Hill; 2004. Acid maltase deficiency.
-
- Ausems MG, Verbiest J, Hermans MP, Kroos MA, Beemer FA, Wokke JH, Sandkuijl LA, Reuser AJ, van der Ploeg AT. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet. 1999;7:713–716. doi: 10.1038/sj.ejhg.5200367. - DOI - PubMed
-
- Martiniuk F, Chen A, Mack A, Arvanitopoulos E, Chen Y, Rom WN, Codd WJ, Hanna B, Alcabes P, Raben N, Plotz P. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet. 1998;79:69–72. doi: 10.1002/(SICI)1096-8628(19980827)79:1<69::AID-AJMG16>3.0.CO;2-K. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
