

Inhibition of PKCδ reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer
- PMID: 21633170
- PMCID: PMC3223835
- DOI: 10.1172/JCI45586
Inhibition of PKCδ reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer
Abstract
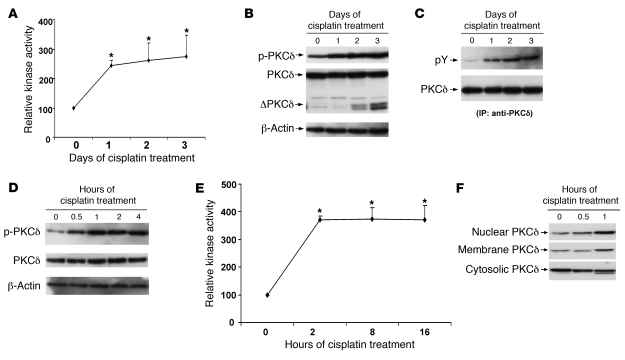
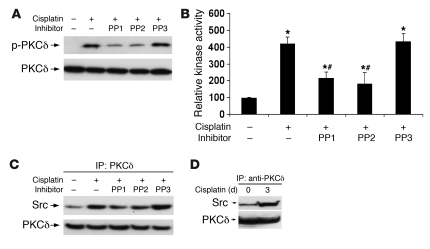
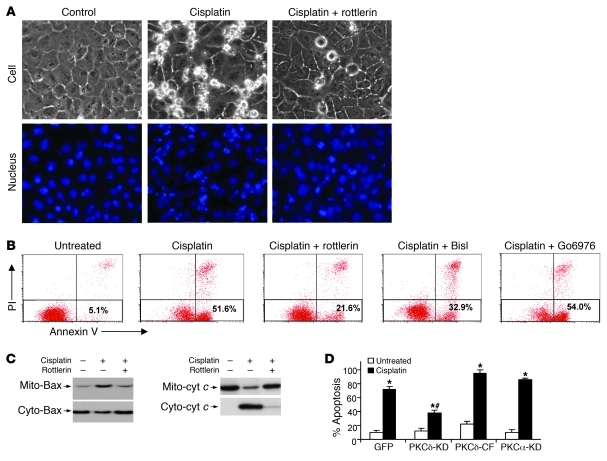
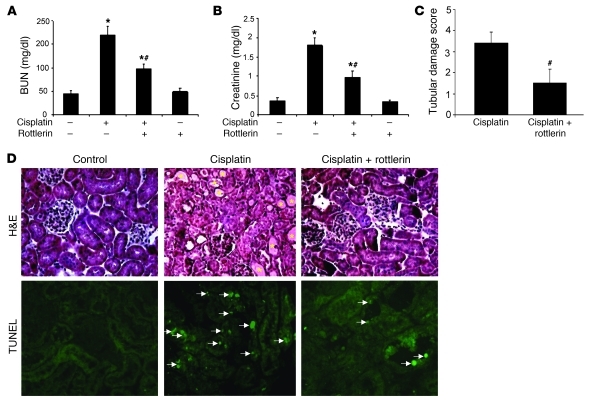
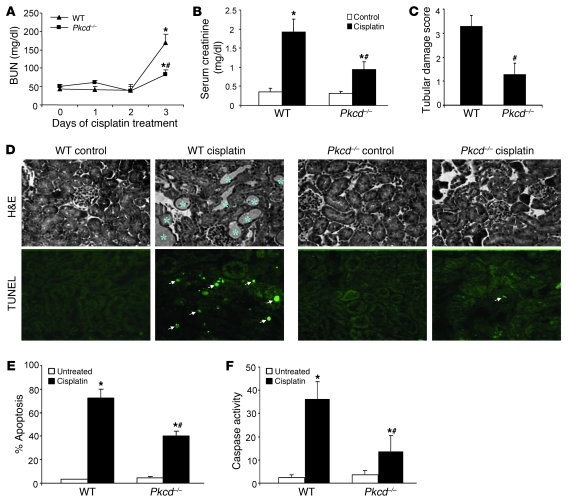
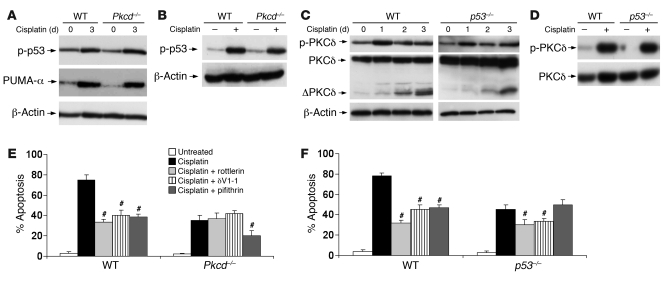
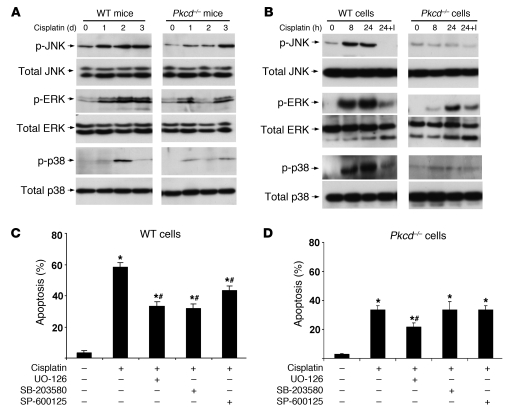
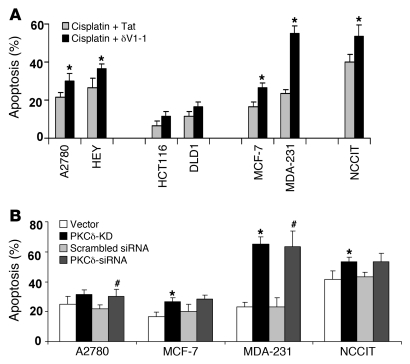
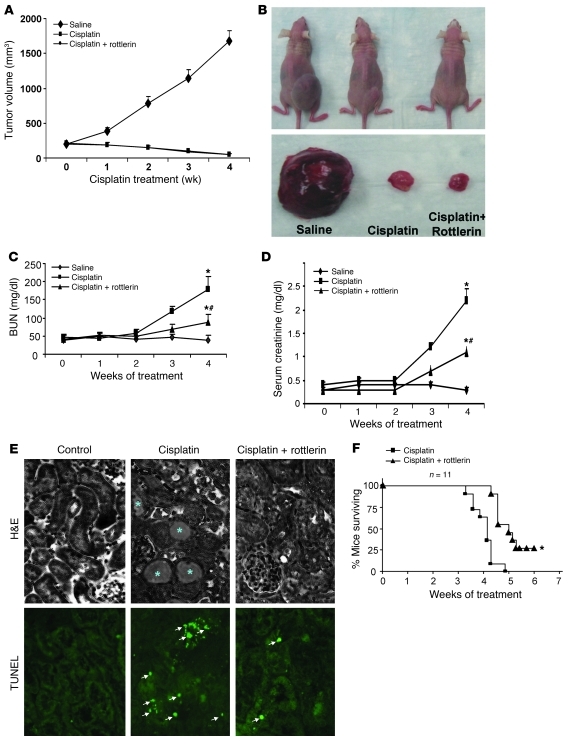
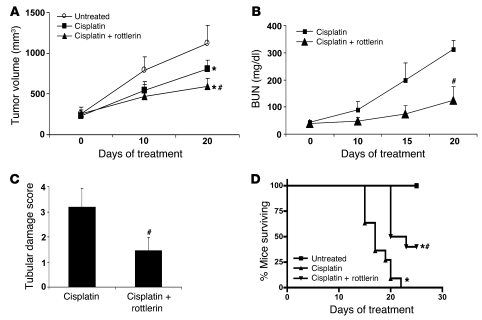
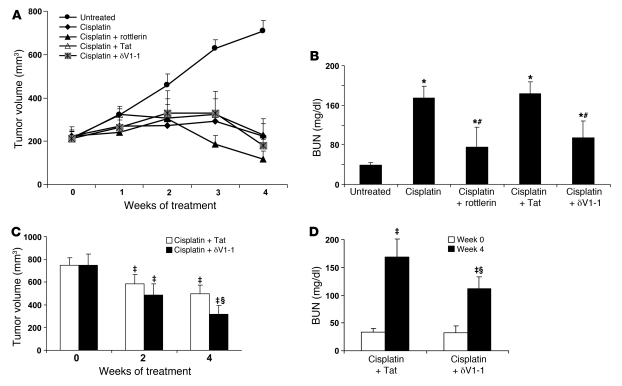
Cisplatin is a widely used cancer therapy drug that unfortunately has major side effects in normal tissues, notably nephrotoxicity in kidneys. Despite intensive research, the mechanism of cisplatin-induced nephrotoxicity remains unclear, and renoprotective approaches during cisplatin-based chemotherapy are lacking. Here we have identified PKCδ as a critical regulator of cisplatin nephrotoxicity, which can be effectively targeted for renoprotection during chemotherapy. We showed that early during cisplatin nephrotoxicity, Src interacted with, phosphorylated, and activated PKCδ in mouse kidney lysates. After activation, PKCδ regulated MAPKs, but not p53, to induce renal cell apoptosis. Thus, inhibition of PKCδ pharmacologically or genetically attenuated kidney cell apoptosis and tissue damage, preserving renal function during cisplatin treatment. Conversely, inhibition of PKCδ enhanced cisplatin-induced cell death in multiple cancer cell lines and, remarkably, enhanced the chemotherapeutic effects of cisplatin in several xenograft and syngeneic mouse tumor models while protecting kidneys from nephrotoxicity. Together these results demonstrate a role of PKCδ in cisplatin nephrotoxicity and support targeting PKCδ as an effective strategy for renoprotection during cisplatin-based cancer therapy.
Figures
References
-
- Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22(47):7265–7279. - PubMed
-
- Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4(4):307–320. - PubMed
-
- Cepeda V, Fuertes MA, Castilla J, Alonso C, Quevedo C, Perez JM. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med Chem. 2007;7(1):3–18. - PubMed
-
- Launay-Vacher V, Rey JB, Isnard-Bagnis C, Deray G, Daouphars M. Prevention of cisplatin nephrotoxicity: state of the art and recommendations from the European Society of Clinical Pharmacy Special Interest Group on Cancer Care. Cancer Chemother Pharmacol. 2008;61(6):903–909. doi: 10.1007/s00280-008-0711-0. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
