

doi: 10.1002/0471250953.bi0214s34.
Using MACS to identify peaks from ChIP-Seq data
Affiliations
- PMID: 21633945
- PMCID: PMC3120977
- DOI: 10.1002/0471250953.bi0214s34
Item in Clipboard
Using MACS to identify peaks from ChIP-Seq data
Curr Protoc Bioinformatics.
2011 Jun.
Abstract
Model-based Analysis of ChIP-Seq (MACS) is a command-line tool designed by X. Shirley Liu and colleagues to analyze data generated by ChIP-Seq experiments in eukaryotes, especially mammals. MACS can be used to identify transcription factor binding sites and histone modification-enriched regions if the ChIP-Seq data, with or without control samples, are given. This unit describes two basic protocols that provide detailed information on how to use MACS to identify either the binding sites of a transcription factor or the enriched regions of a histone modification with broad peaks. Furthermore, the basic ideas for the MACS algorithm and its appropriate usage are discussed.
Figures
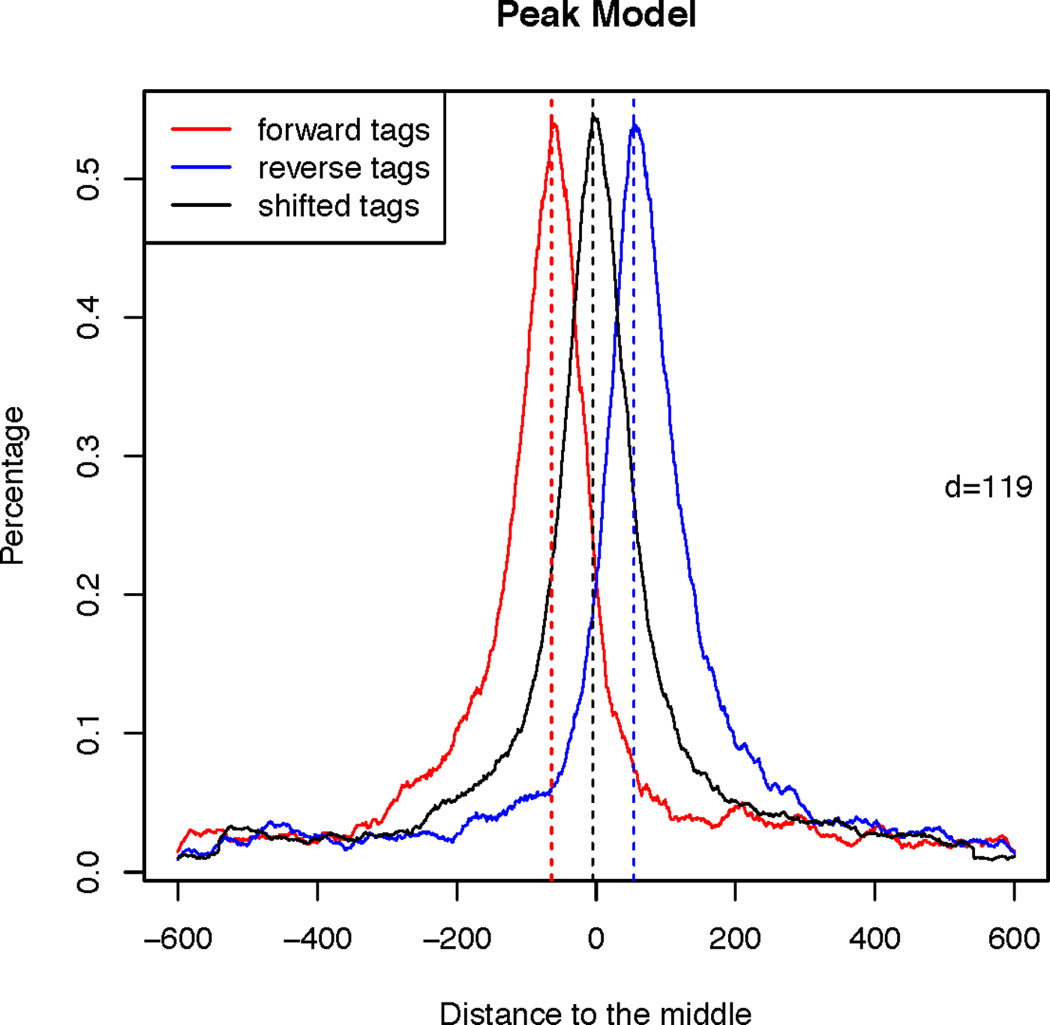
Shifting size model for FoxA1 ChIP-Seq data.
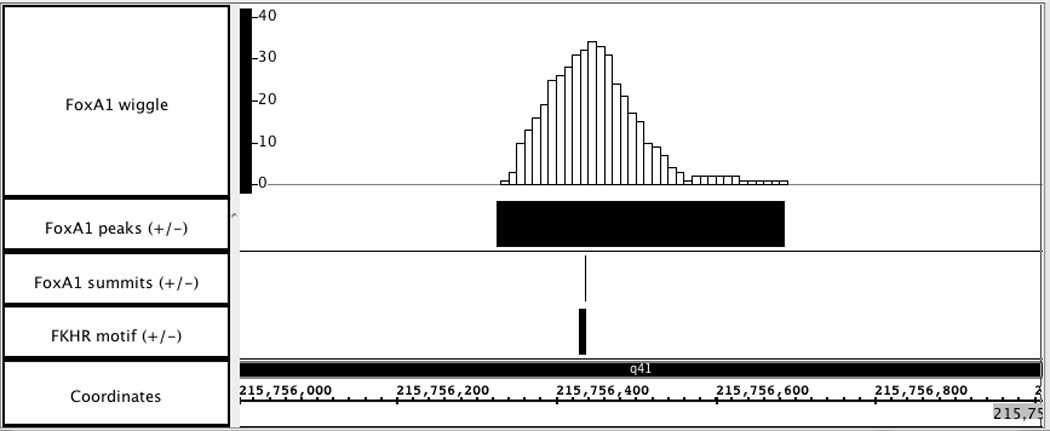
Visualization of wiggle and BED files for FoxA1 ChIP-Seq data in Affymetrix IGB. Genome region: chr1: 215,756,000 – 215,757,000.
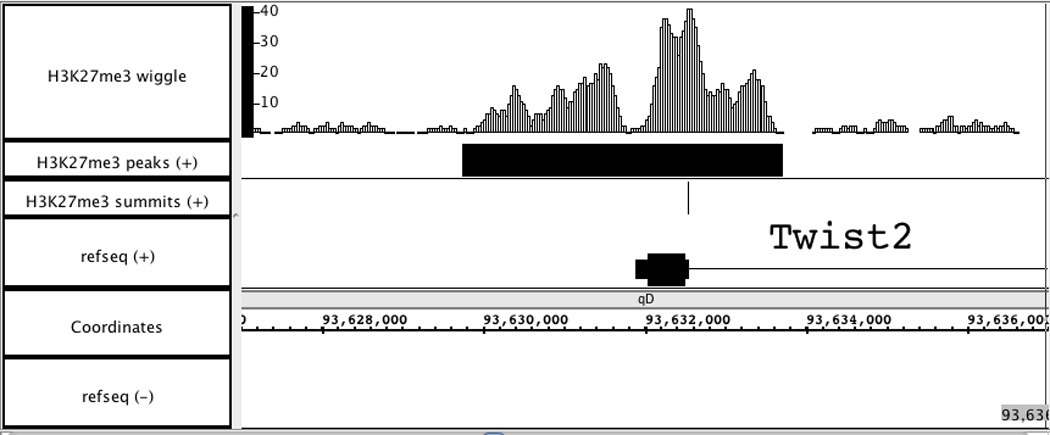
Visualization of wiggle and BED files for H3K27me3 ChIP-Seq data in Affymetrix IGB. Genome region: chr1: 93,627,000 – 93,637,000.
References
-
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. - PubMed
-
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. - PubMed
-
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
