

Machado-Joseph Disease: from first descriptions to new perspectives
- PMID: 21635785
- PMCID: PMC3123549
- DOI: 10.1186/1750-1172-6-35
Machado-Joseph Disease: from first descriptions to new perspectives
Abstract
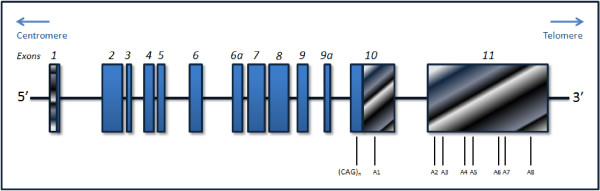
Machado-Joseph Disease (MJD), also known as spinocerebellar ataxia type 3 (SCA3), represents the most common form of SCA worldwide. MJD is an autosomal dominant neurodegenerative disorder of late onset, involving predominantly the cerebellar, pyramidal, extrapyramidal, motor neuron and oculomotor systems; although sharing features with other SCAs, the identification of minor, but more specific signs, facilitates its differential diagnosis. MJD presents strong phenotypic heterogeneity, which has justified the classification of patients into three main clinical types. Main pathological lesions are observed in the spinocerebellar system, as well as in the cerebellar dentate nucleus. MJD's causative mutation consists in an expansion of an unstable CAG tract in exon 10 of the ATXN3 gene, located at 14q32.1. Haplotype-based studies have suggested that two main founder mutations may explain the present global distribution of the disease; the ancestral haplotype is of Asian origin, and has an estimated age of around 5,800 years, while the second mutational event has occurred about 1,400 years ago. The ATXN3 gene encodes for ataxin-3, which is ubiquitously expressed in neuronal and non-neuronal tissues, and, among other functions, is thought to participate in cellular protein quality control pathways. Mutated ATXN3 alleles consensually present about 61 to 87 CAG repeats, resulting in an expanded polyglutamine tract in ataxin-3. This altered protein gains a neurotoxic function, through yet unclear mechanisms. Clinical variability of MJD is only partially explained by the size of the CAG tract, which leaves a residual variance that should be explained by still unknown additional factors. Several genetic tests are available for MJD, and Genetic Counseling Programs have been created to better assist the affected families, namely on what concerns the possibility of pre-symptomatic testing. The main goal of this review was to bring together updated knowledge on MJD, covering several aspects from its initial descriptions and clinical presentation, through the discovery of the causative mutation, its origin and dispersion, as well as molecular genetics aspects considered essential for a better understanding of its neuropathology. Issues related with molecular testing and Genetic Counseling, as well as recent progresses and perspectives on genetic therapy, are also addressed.
Figures
References
-
- Coutinho P, Andrade C. Autosomal dominant system degeneration in Portuguese families of the Azores Islands. A new genetic disorder involving cerebellar, pyramidal, extrapyramidal and spinal cord motor functions. Neurology. 1978;28(7):703–709. - PubMed
-
- Stevanin G, Le Guern E, Ravise N, Chneiweiss H, Durr A, Cancel G, Vignal A, Boch AL, Ruberg M, Penet C. et al. A third locus for autosomal dominant cerebellar ataxia type I maps to chromosome 14q24.3-qter: evidence for the existence of a fourth locus. Am J Hum Genet. 1994;54(1):11–20. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
