

The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease
- PMID: 21637107
- PMCID: PMC3954622
- DOI: 10.1097/GIM.0b013e3182174703
The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease
Abstract
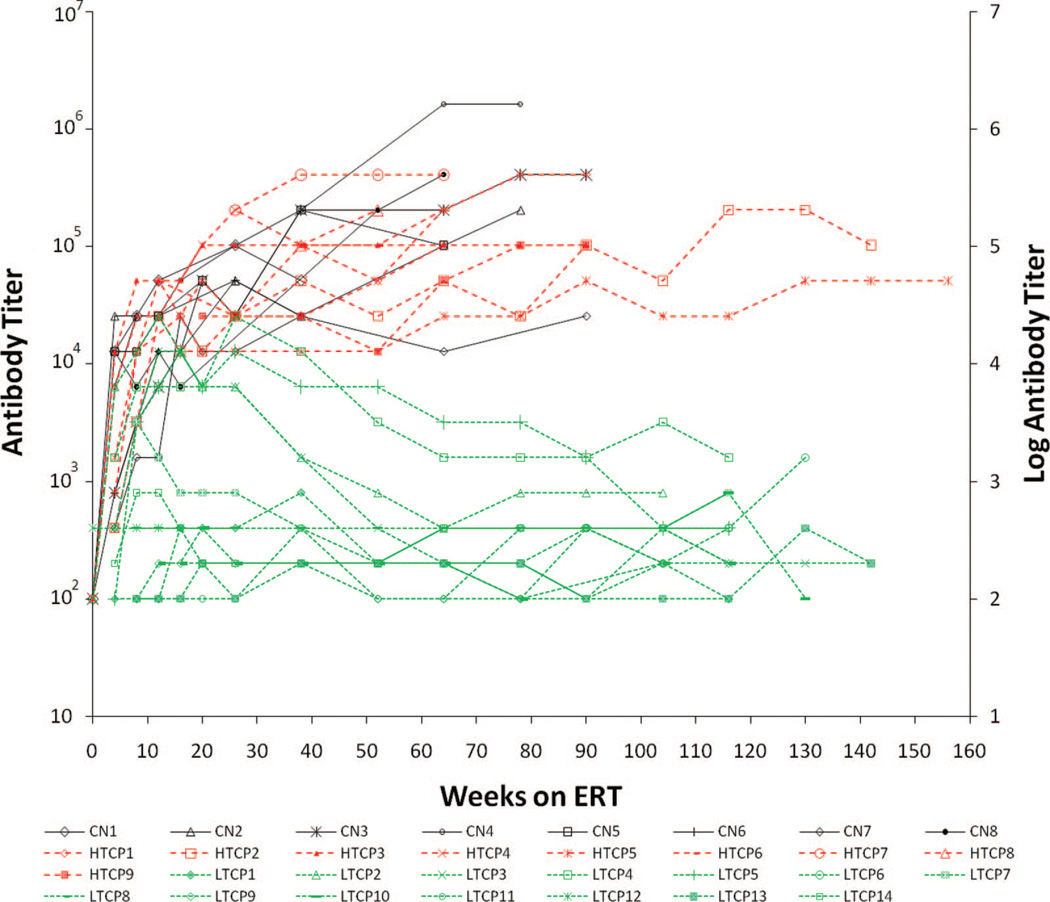
Purpose: Enzyme replacement therapy with rhGAA (Myozyme®) has lead to improved survival, which is largely attributable to improvements in cardiomyopathy and skeletal muscle function. However, crossreactive immunologic material-negative patients have a poor clinical response to enzyme replacement therapy secondary to high sustained antibody titers. Furthermore, although the majority of crossreactive immunologic material-positive patients tolerize or experience a downtrend in anti-rhGAA antibody titers, antibody response is variable with some crossreactive immunologic material-positive infants also mounting high sustained antibody titers.
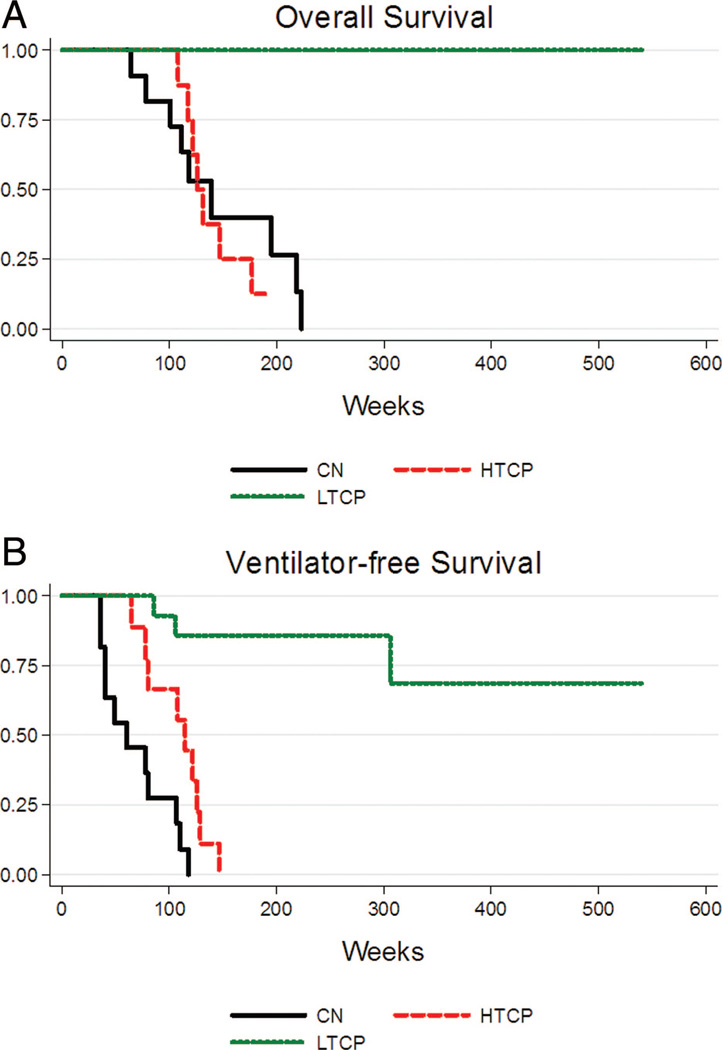
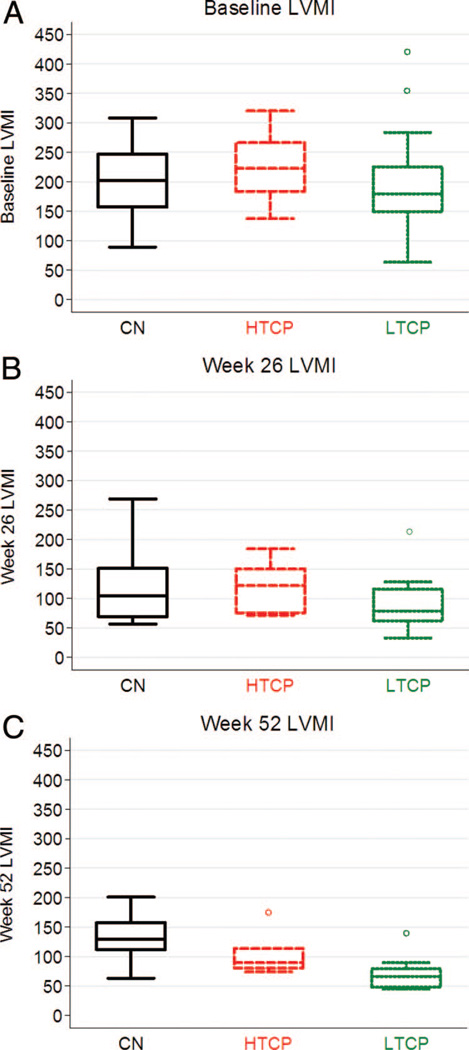
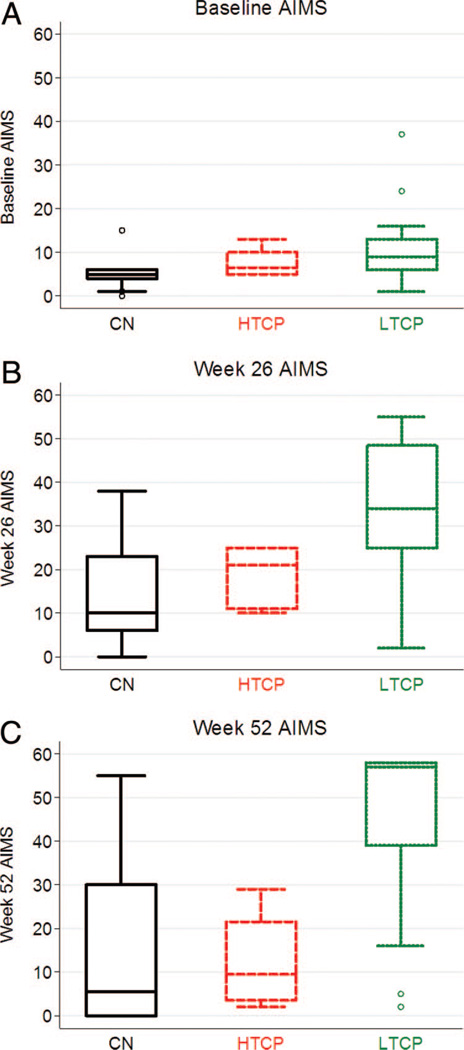
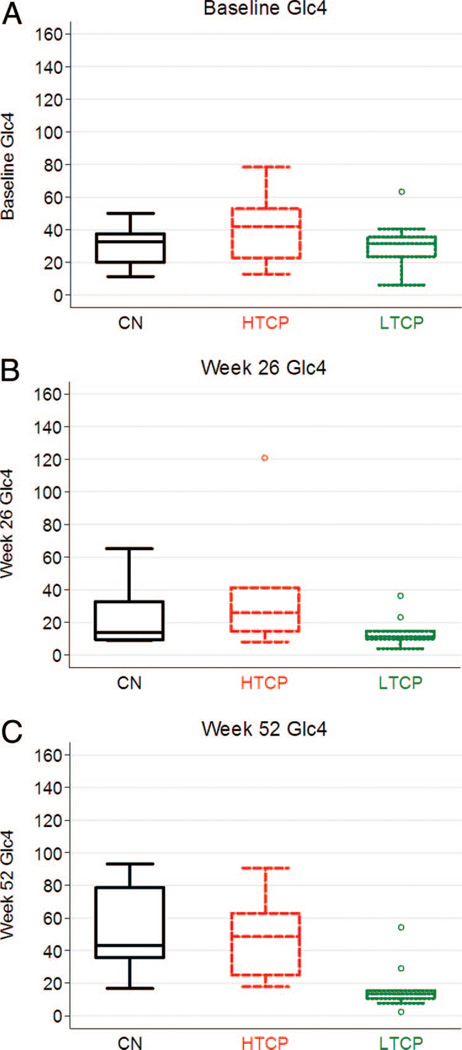
Methods: We retrospectively analyzed 34 infants with Pompe disease: 11 crossreactive immunologic material-negative patients, nine high-titer crossreactive immunologic material-positive patients, and 14 low-titer crossreactive immunologic material-positive patients. Clinical outcome measures were overall survival, ventilator-free survival, left ventricular mass index, Alberta Infant Motor Scale score, and urine Glc(4) levels.
Results: Clinical outcomes in the high-titer crossreactive immunologic material-positive group were poor across all areas evaluated relative to the low-titer crossreactive immunologic material-positive group. For the crossreactive immunologic material-negative and high-titer crossreactive immunologic material-positive groups, no statistically significant differences were observed for any outcome measures, and both patient groups did poorly.
Conclusions: Our data indicate that, irrespective of crossreactive immunologic material status, patients with infantile Pompe disease with high sustained antibody titer have an attenuated therapeutic response to enzyme replacement therapy. With the advent of immunomodulation therapies, identification of patients at risk for developing high sustained antibody titer is critical.
Conflict of interest statement
This potential conflict for Duke University has been resolved through monetization. S. G. Banugaria, S. N. Prater, Y-K. Ng, J. A. Kobori, R.S. Finkel, R. L. Ladda and A. S. Rosenberg have no financial or proprietary interest in the materials presented herein.
Figures
References
-
- Hirschhorn R, Reuser AJJ. New York: McGraw-Hill; 2009. Glycogen storage disease type II: acid a-glucosidase (acid maltase) deficiency [Internet Resource; Computer File Date of Entry: 20090331] Available at: http://genetics.accessmedicine.com/.
-
- Hug G. Pre- and postnatal pathology, enzyme treatment, and unresolved issues in five lysosomal disorders. Pharmacol Rev. 1978;30:565–591. - PubMed
-
- van den Hout HM, Hop W, van Diggelen OP, et al. The natural course of infantile Pompe’s disease, 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332–340. - PubMed
-
- Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148:671–676. - PubMed
-
- Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99–109. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
