

SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis
- PMID: 21637290
- PMCID: PMC3170683
- DOI: 10.1038/cdd.2011.71
SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis
Abstract
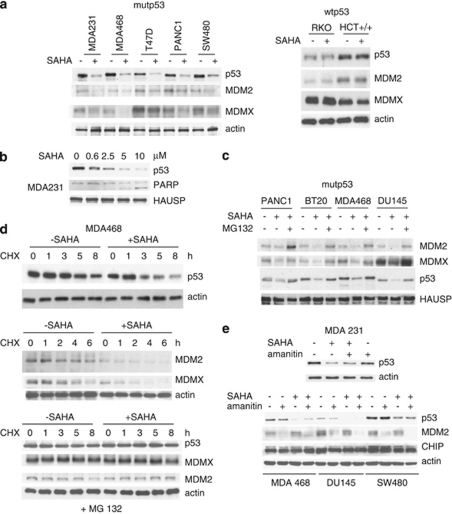
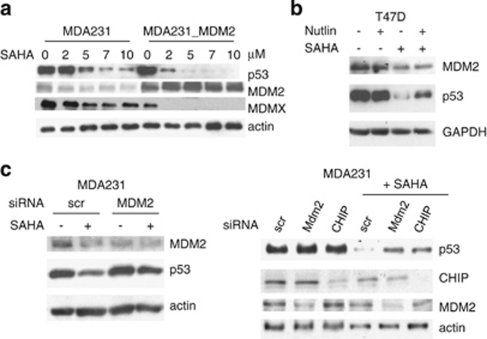
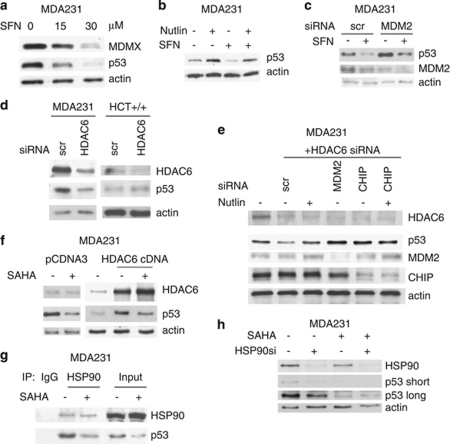
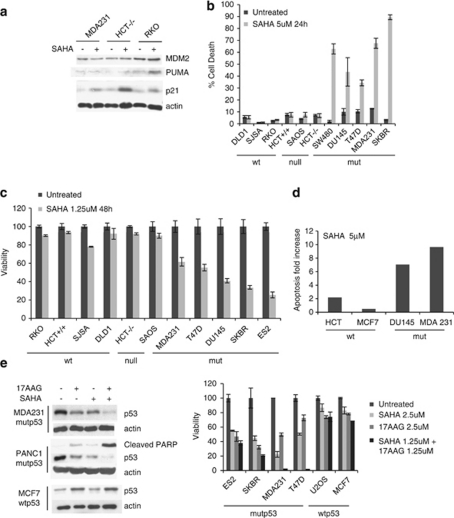
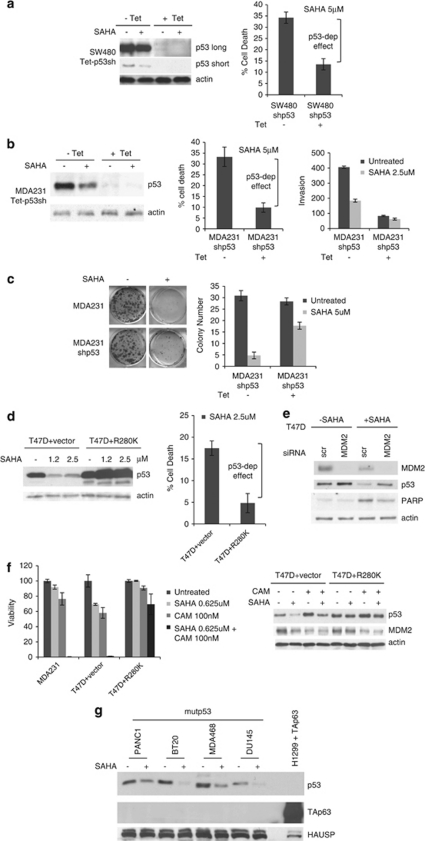
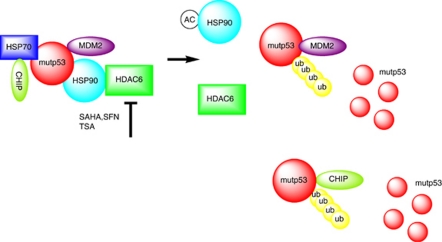
Mutant p53 (mutp53) cancers are surprisingly dependent on their hyperstable mutp53 protein for survival, identifying mutp53 as a potentially significant clinical target. However, exploration of effective small molecule therapies targeting mutp53 has barely begun. Mutp53 hyperstabilization, a hallmark of p53 mutation, is cancer cell-specific and due to massive upregulation of the HSP90 chaperone machinery during malignant transformation. We recently showed that stable complex formation between HSP90 and its mutp53 client inhibits E3 ligases MDM2 and CHIP, causing mutp53 stabilization. Histone deacetylase (HDAC) inhibitors (HDACi) are a new class of promising anti-cancer drugs, hyperacetylating histone and non-histone targets. Currently, suberoylanilide hydroxamic acid (SAHA) is the only FDA-approved HDACi. We show that SAHA exhibits preferential cytotoxicity for mutant, rather than wild-type and null p53 human cancer cells. Loss/gain-of-function experiments revealed that although able to exert multiple cellular effects, SAHA's cytotoxicity is caused to a significant degree by its ability to strongly destabilize mutp53 at the level of protein degradation. The underlying mechanism is SAHA's inhibition of HDAC6, an essential positive regulator of HSP90. This releases mutp53 and enables its MDM2- and CHIP-mediated degradation. SAHA also strongly chemosensitizes mutp53 cancer cells for chemotherapy due to its ability to degrade mutp53. This identifies a novel action of SAHA with the prospect of SAHA becoming a centerpiece in mutp53-specific anticancer strategies.
Figures
References
-
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872. - PubMed
-
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. - PubMed
-
- Blandino G, Levine AJ, Oren M. Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene. 1999;18:477–485. - PubMed
-
- Li R, Sutphin PD, Schwartz D, Matas D, Almog N, Wolkowicz R, et al. Mutant p53 protein expression interferes with p53-independent apoptotic pathways. Oncogene. 1998;16:3269–3277. - PubMed
-
- Bossi G, Lapi E, Strano S, Rinaldo C, Blandino G, Sacchi A. Mutant p53 gain of function: reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene. 2006;25:304–309. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
