

Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation
- PMID: 21637795
- PMCID: PMC3102752
- DOI: 10.1371/journal.pcbi.1002061
Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation
Abstract
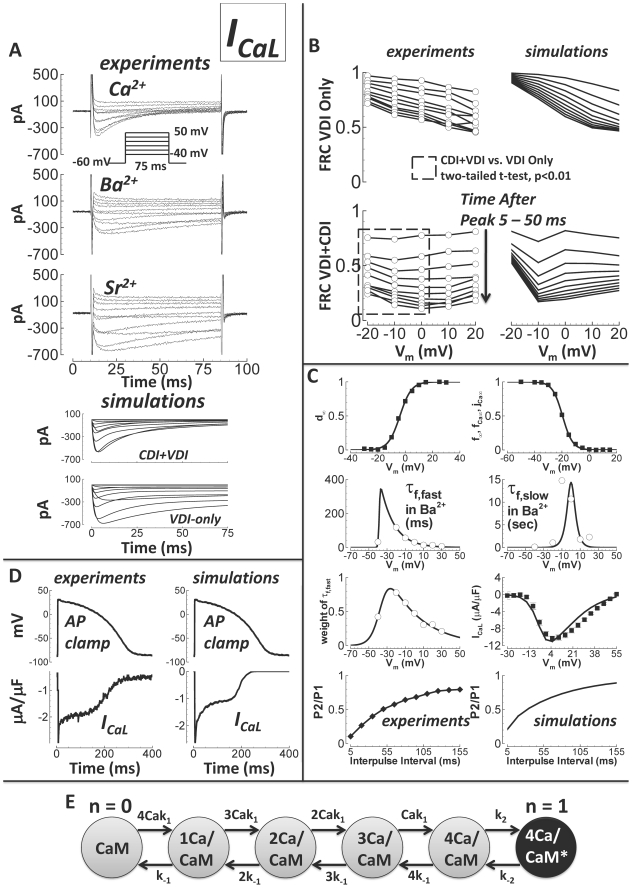
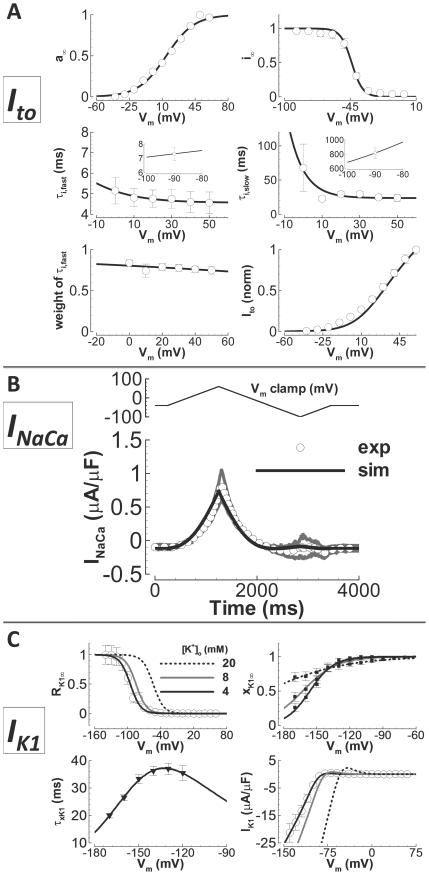
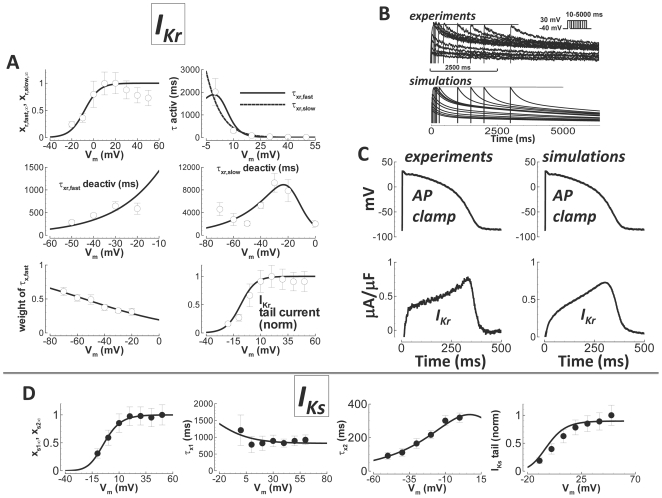
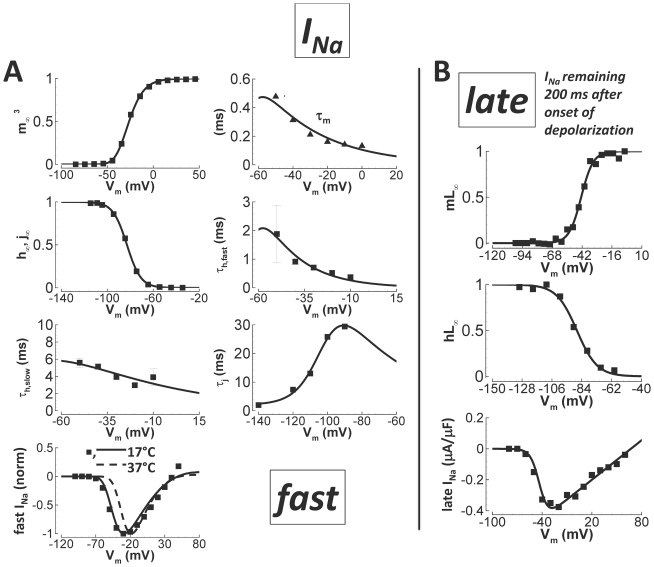
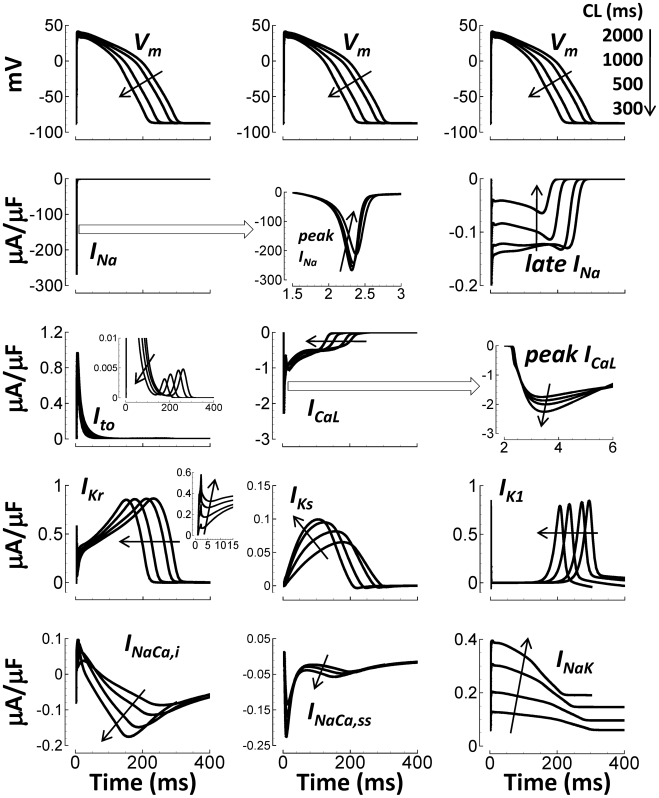
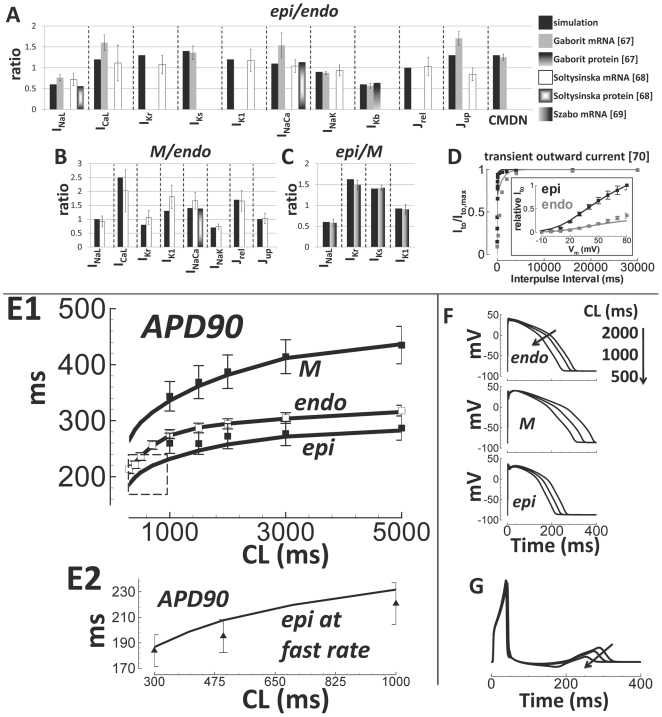
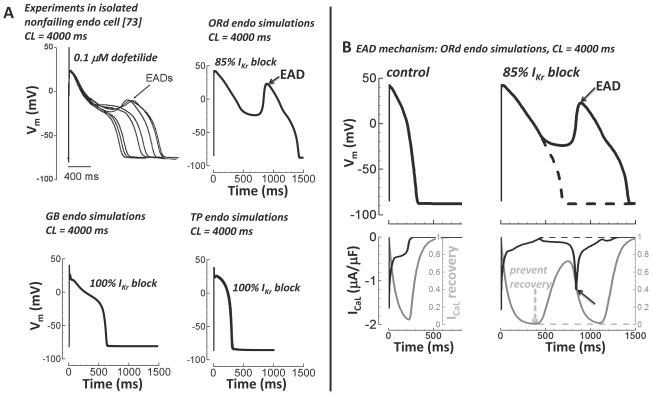
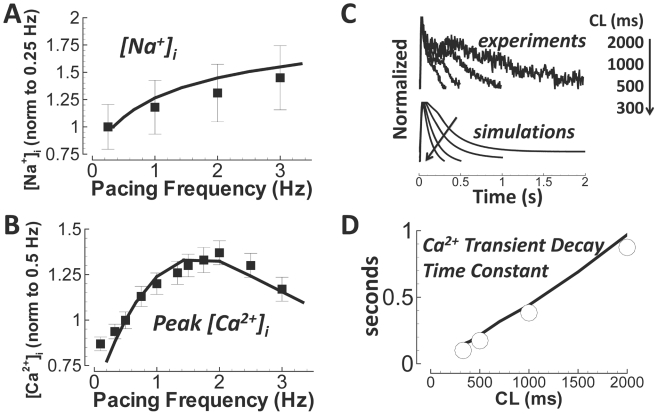
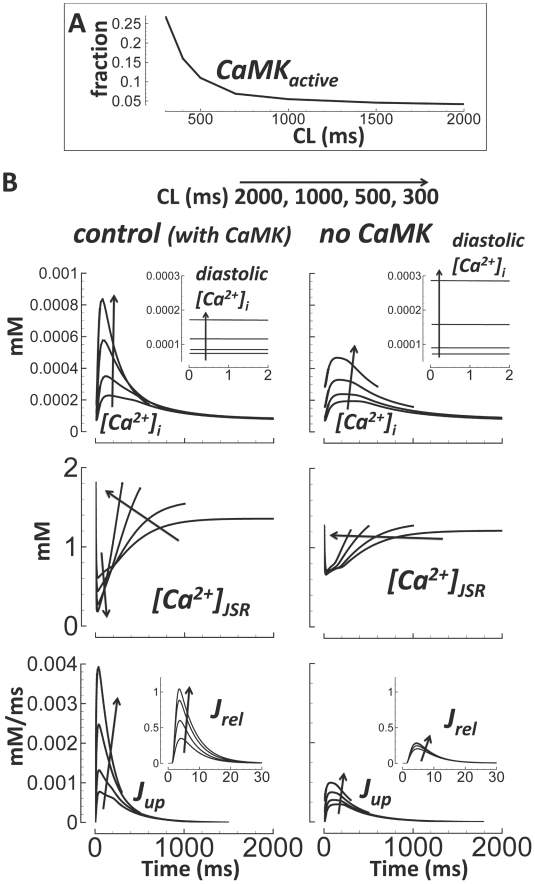
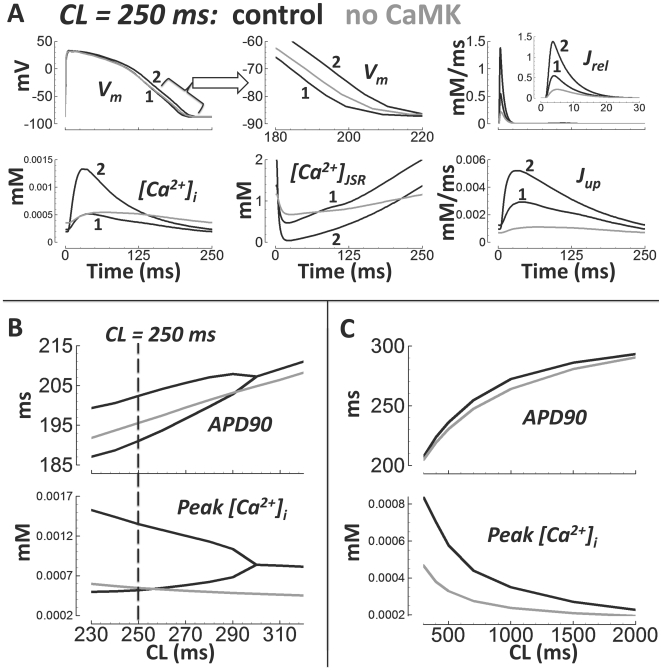
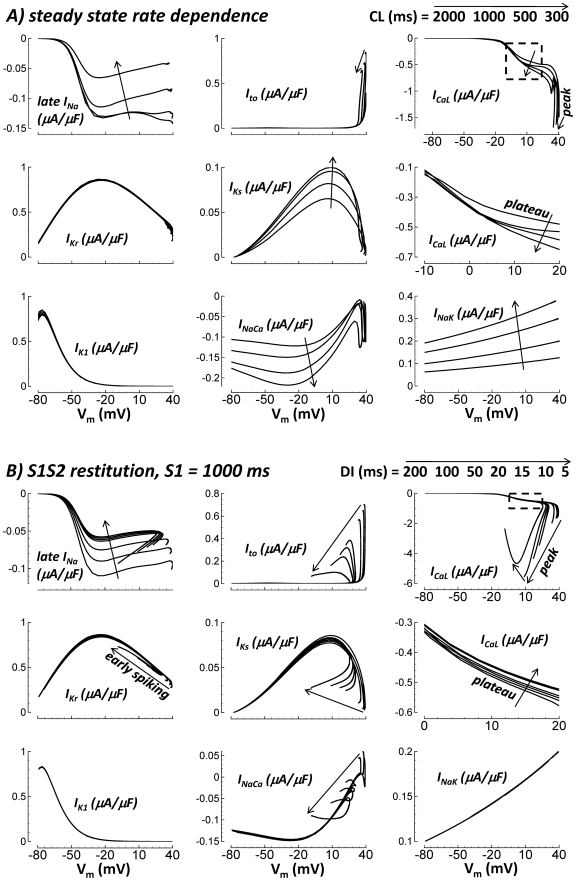
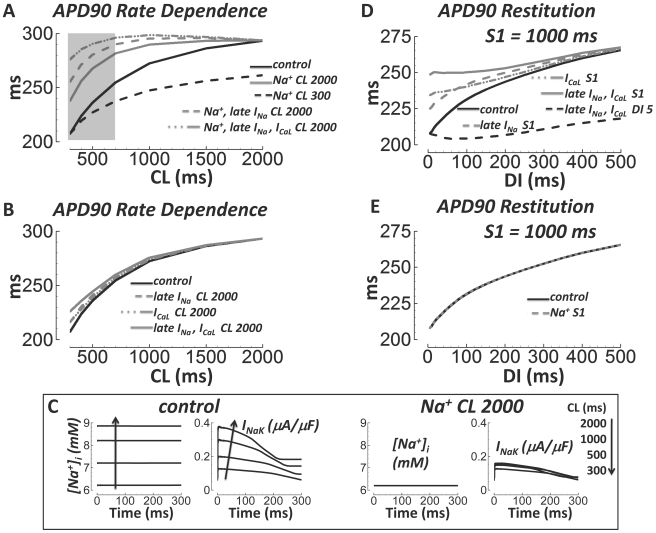
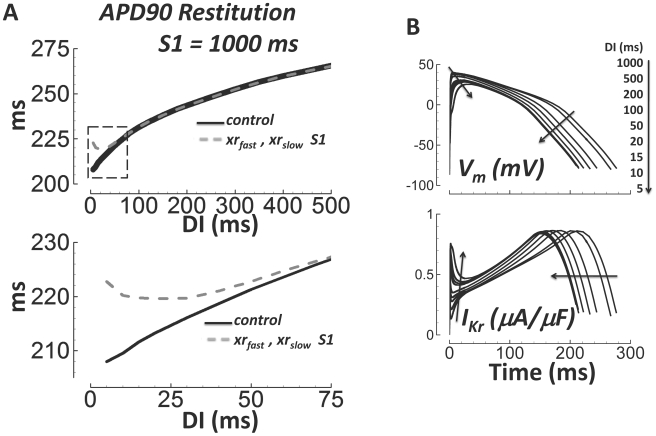
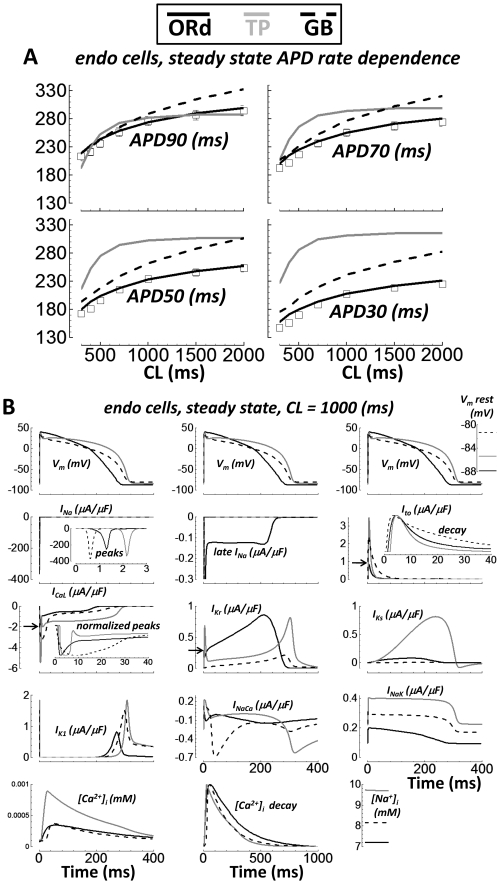
Cellular electrophysiology experiments, important for understanding cardiac arrhythmia mechanisms, are usually performed with channels expressed in non myocytes, or with non-human myocytes. Differences between cell types and species affect results. Thus, an accurate model for the undiseased human ventricular action potential (AP) which reproduces a broad range of physiological behaviors is needed. Such a model requires extensive experimental data, but essential elements have been unavailable. Here, we develop a human ventricular AP model using new undiseased human ventricular data: Ca(2+) versus voltage dependent inactivation of L-type Ca(2+) current (I(CaL)); kinetics for the transient outward, rapid delayed rectifier (I(Kr)), Na(+)/Ca(2+) exchange (I(NaCa)), and inward rectifier currents; AP recordings at all physiological cycle lengths; and rate dependence and restitution of AP duration (APD) with and without a variety of specific channel blockers. Simulated APs reproduced the experimental AP morphology, APD rate dependence, and restitution. Using undiseased human mRNA and protein data, models for different transmural cell types were developed. Experiments for rate dependence of Ca(2+) (including peak and decay) and intracellular sodium ([Na(+)](i)) in undiseased human myocytes were quantitatively reproduced by the model. Early afterdepolarizations were induced by I(Kr) block during slow pacing, and AP and Ca(2+) alternans appeared at rates >200 bpm, as observed in the nonfailing human ventricle. Ca(2+)/calmodulin-dependent protein kinase II (CaMK) modulated rate dependence of Ca(2+) cycling. I(NaCa) linked Ca(2+) alternation to AP alternans. CaMK suppression or SERCA upregulation eliminated alternans. Steady state APD rate dependence was caused primarily by changes in [Na(+)](i), via its modulation of the electrogenic Na(+)/K(+) ATPase current. At fast pacing rates, late Na(+) current and I(CaL) were also contributors. APD shortening during restitution was primarily dependent on reduced late Na(+) and I(CaL) currents due to inactivation at short diastolic intervals, with additional contribution from elevated I(Kr) due to incomplete deactivation.
Conflict of interest statement
The authors have declared that no competing interests exist.
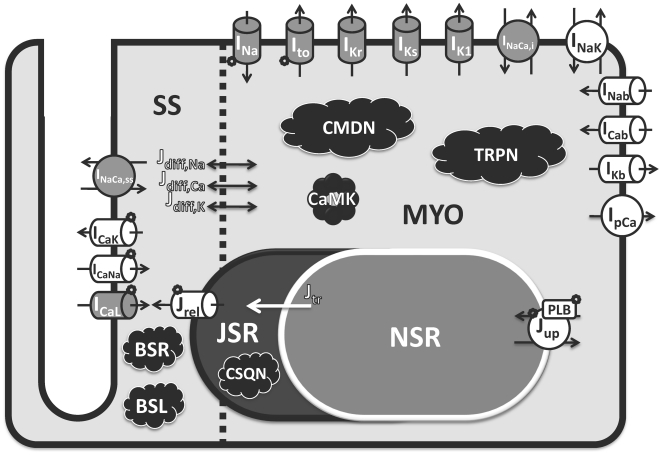
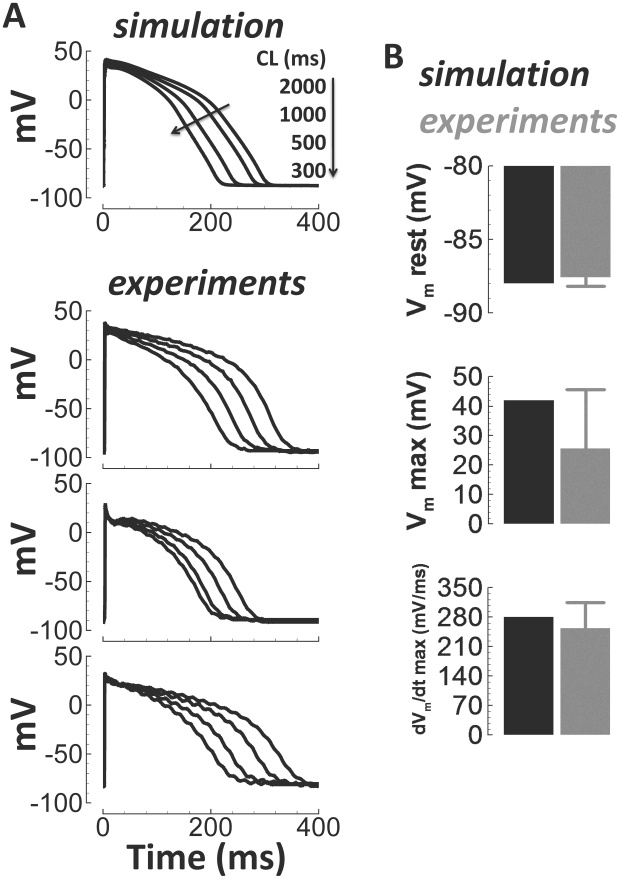
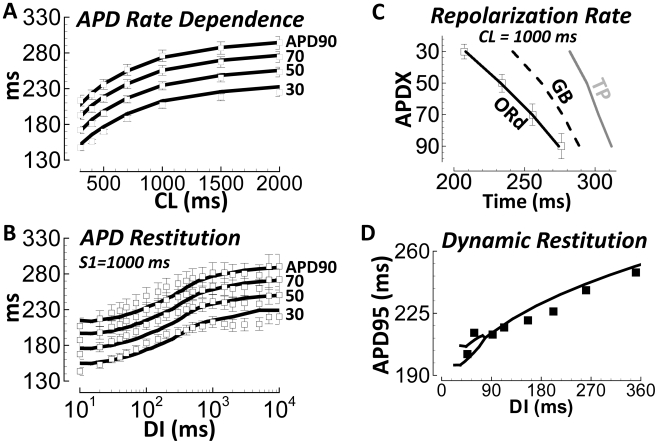
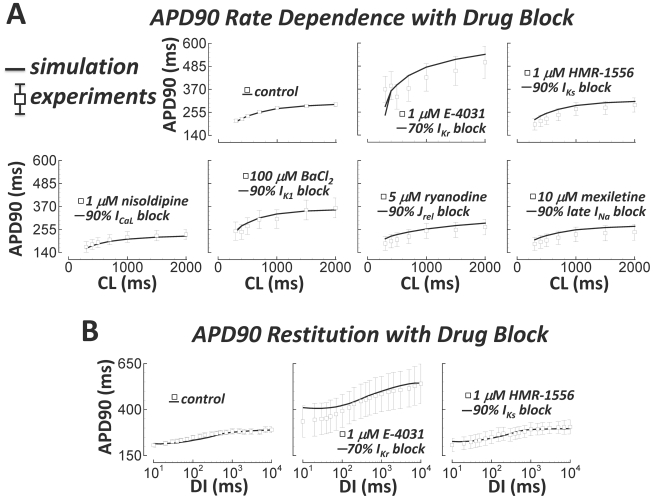
Figures
References
-
- Myerburg RJ. Scientific gaps in the prediction and prevention of sudden cardiac death. J Cardiovasc Electrophysiol. 2002;13:709–723. - PubMed
-
- Akar FG, Wu RC, Deschenes I, Armoundas AA, Piacentino V, 3rd, et al. Phenotypic differences in transient outward K+ current of human and canine ventricular myocytes: insights into molecular composition of ventricular Ito. Am J Physiol Heart Circ Physiol. 2004;286:H602–609. - PubMed
-
- Blechschmidt S, Haufe V, Benndorf K, Zimmer T. Voltage-gated Na+ channel transcript patterns in the mammalian heart are species-dependent. Prog Biophys Mol Biol. 2008;98:309–318. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
