

Incorporation of evolutionary information into Rosetta comparative modeling
- PMID: 21638331
- PMCID: PMC3538865
- DOI: 10.1002/prot.23046
Incorporation of evolutionary information into Rosetta comparative modeling
Abstract
Prediction of protein structures from sequences is a fundamental problem in computational biology. Algorithms that attempt to predict a structure from sequence primarily use two sources of information. The first source is physical in nature: proteins fold into their lowest energy state. Given an energy function that describes the interactions governing folding, a method for constructing models of protein structures, and the amino acid sequence of a protein of interest, the structure prediction problem becomes a search for the lowest energy structure. Evolution provides an orthogonal source of information: proteins of similar sequences have similar structure, and therefore proteins of known structure can guide modeling. The relatively successful Rosetta approach takes advantage of the first, but not the second source of information during model optimization. Following the classic work by Andrej Sali and colleagues, we develop a probabilistic approach to derive spatial restraints from proteins of known structure using advances in alignment technology and the growth in the number of structures in the Protein Data Bank. These restraints define a region of conformational space that is high-probability, given the template information, and we incorporate them into Rosetta's comparative modeling protocol. The combined approach performs considerably better on a benchmark based on previous CASP experiments. Incorporating evolutionary information into Rosetta is analogous to incorporating sparse experimental data: in both cases, the additional information eliminates large regions of conformational space and increases the probability that energy-based refinement will hone in on the deep energy minimum at the native state.
Copyright © 2011 Wiley-Liss, Inc.
Figures
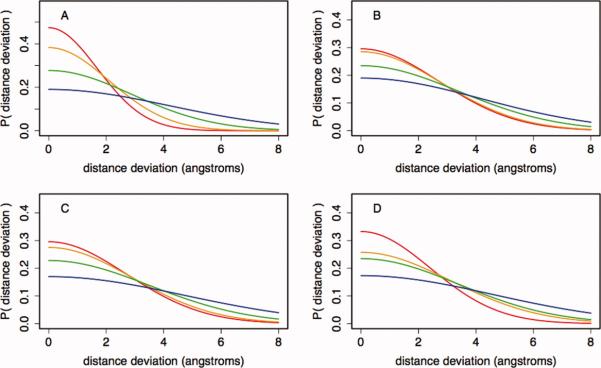
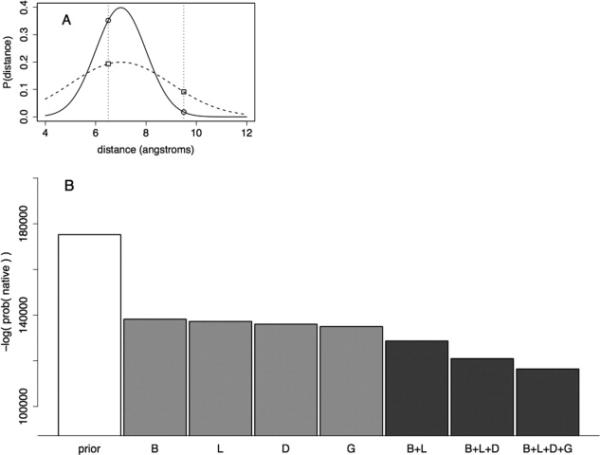
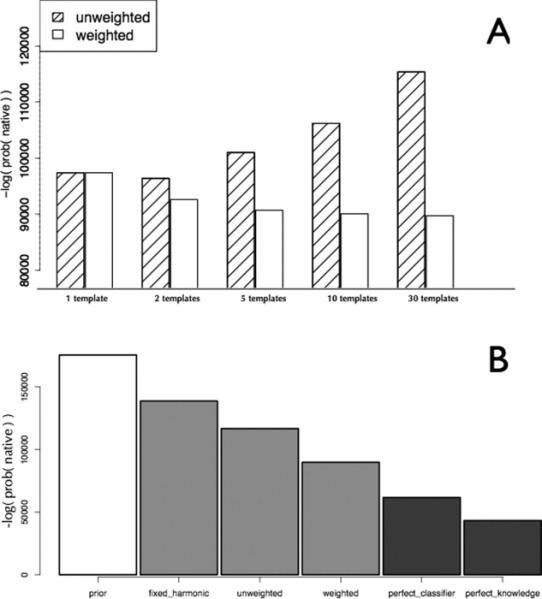
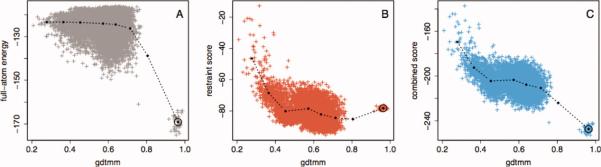
References
-
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. - PubMed
-
- Wang G, Dunbrack RL., Jr PISCES: a protein sequence culling server. Bioinformatics. 2003;19:1589–1591. - PubMed
-
- Söding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
