

Recent advances in the genetics of Parkinson's disease
- PMID: 21639795
- PMCID: PMC4120236
- DOI: 10.1146/annurev-genom-082410-101440
Recent advances in the genetics of Parkinson's disease
Abstract
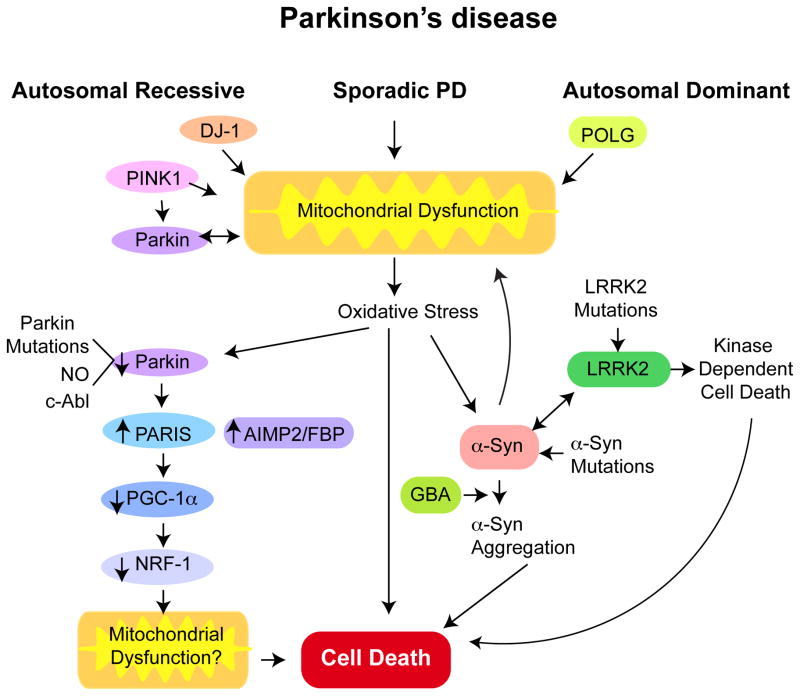
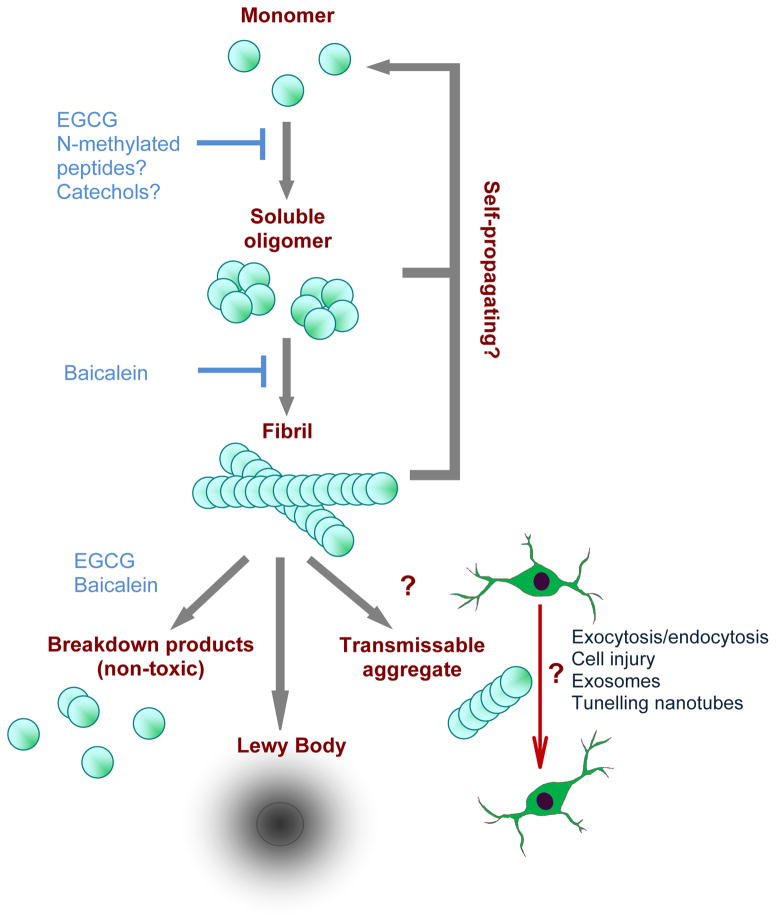
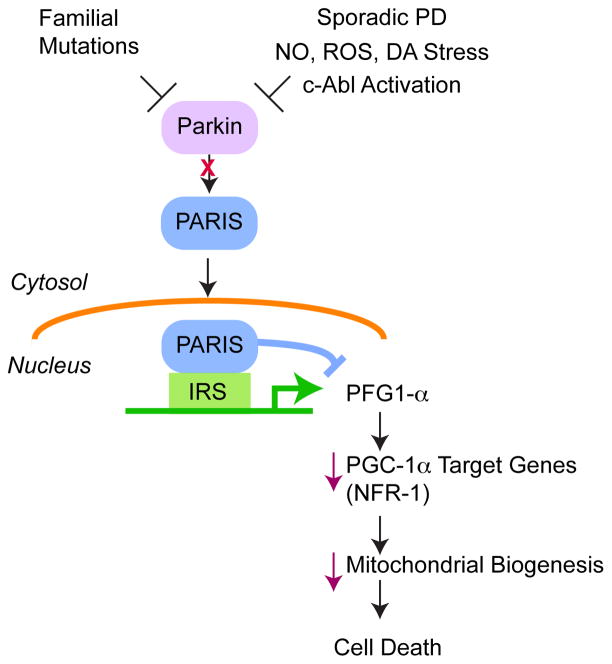
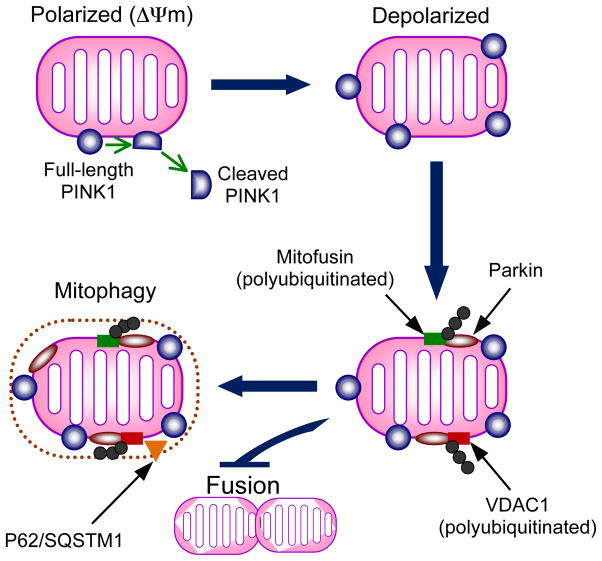
Genetic studies have provided valuable insight into the pathological mechanisms underlying Parkinson's disease (PD). The elucidation of genetic components to what was once largely considered a nongenetic disease has given rise to a multitude of cell and animal models enabling the dissection of molecular pathways involved in disease etiology. Here, we review advances obtained from models of dominant mutations in α-synuclein and LRRK2 as well as recessive PINK1, parkin and DJ-1 mutations. Recent genome-wide association studies have implicated genetic variability at two of these loci, α-synuclein and LRRK2, as significant risk factors for developing sporadic PD. This, coupled with the established role of mitochondrial impairment in both familial and sporadic PD, highlights the likelihood of common mechanisms fundamental to the etiology of both.
Figures

References
-
- Autere J, Moilanen JS, Finnila S, Soininen H, Mannermaa A, et al. Mitochondrial DNA polymorphisms as risk factors for Parkinson’s disease and Parkinson’s disease dementia. Hum Genet. 2004;115:29–35. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
