

Targeting F508del-CFTR to develop rational new therapies for cystic fibrosis
- PMID: 21642944
- PMCID: PMC4009972
- DOI: 10.1038/aps.2011.71
Targeting F508del-CFTR to develop rational new therapies for cystic fibrosis
Abstract
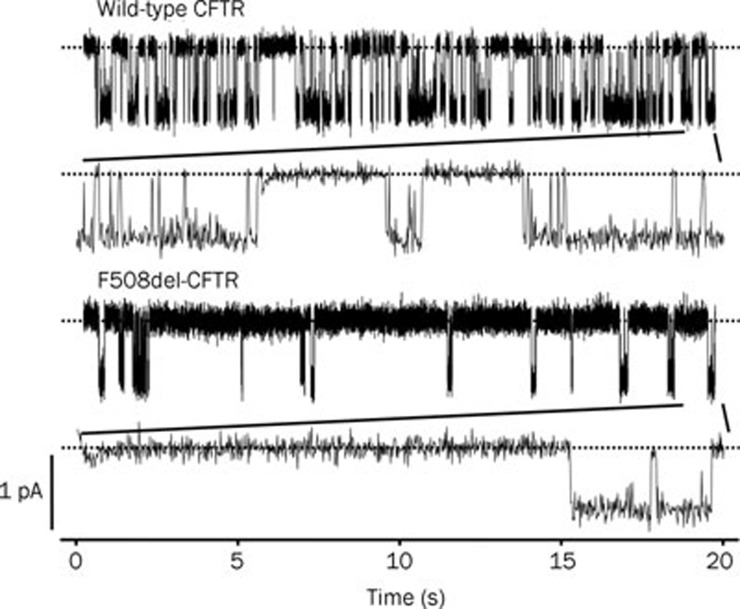
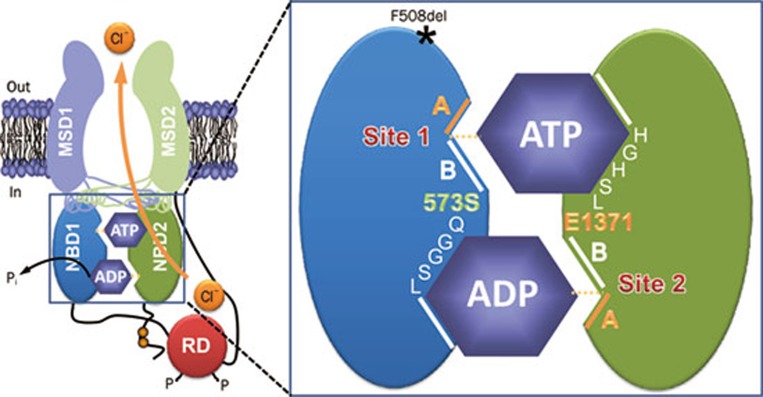
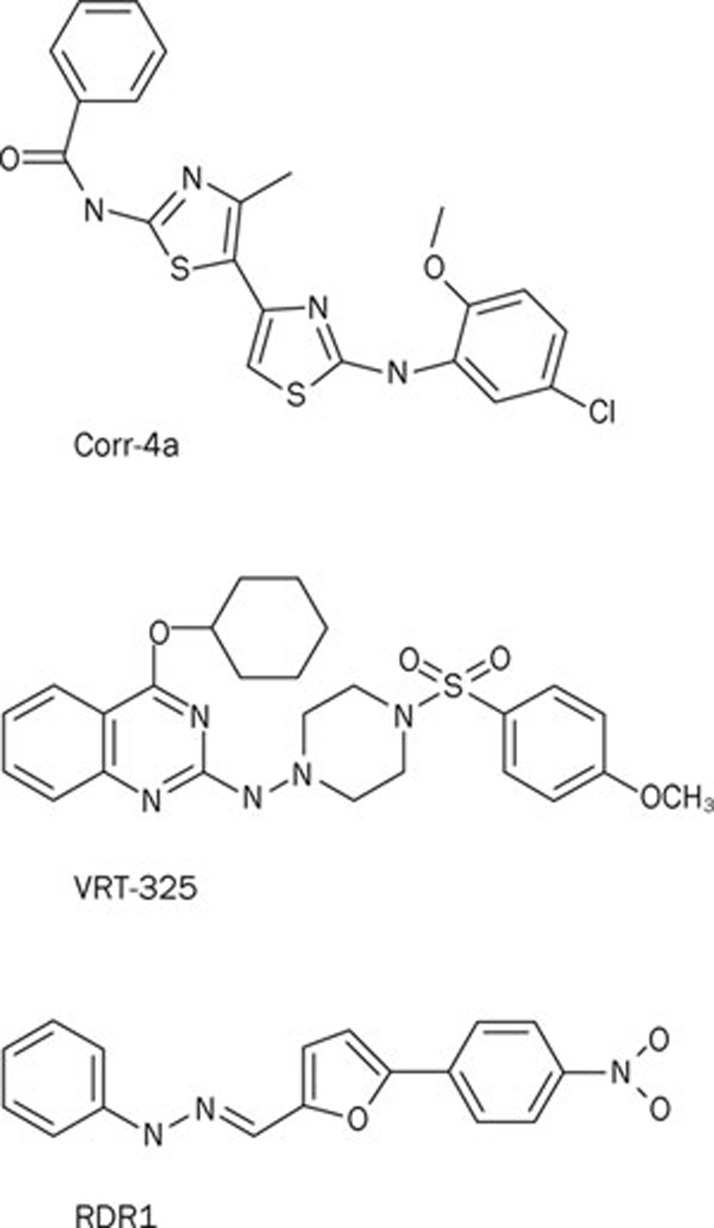
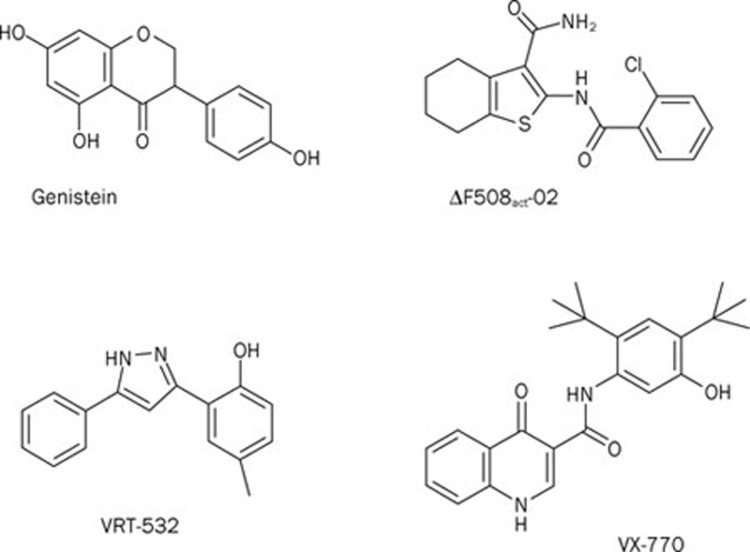
The mutation F508del is the commonest cause of the genetic disease cystic fibrosis (CF). CF disrupts the function of many organs in the body, most notably the lungs, by perturbing salt and water transport across epithelial surfaces. F508del causes harm in two principal ways. First, the mutation prevents delivery of the cystic fibrosis transmembrane conductance regulator (CFTR) to its correct cellular location, the apical (lumen-facing) membrane of epithelial cells. Second, F508del perturbs the Cl(-) channel function of CFTR by disrupting channel gating. Here, we discuss the development of rational new therapies for CF that target F508del-CFTR. We highlight how structural studies provide new insight into the role of F508 in the regulation of channel gating by cycles of ATP binding and hydrolysis. We emphasize the use of high-throughput screening to identify lead compounds for therapy development. These compounds include CFTR correctors that restore the expression of F508del-CFTR at the apical membrane of epithelial cells and CFTR potentiators that rescue the F508del-CFTR gating defect. Initial results from clinical trials of CFTR correctors and potentiators augur well for the development of small molecule therapies that target the root cause of CF: mutations in CFTR.
Figures
References
-
- Welsh MJ, Ramsey BW, Accurso F, Cutting GR.Cystic fibrosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease New York: McGraw-Hill Inc; 2001. p5121–88.
-
- Davis PB. Cystic fibrosis since 1938. Am J Respir Crit Care Med. 2006;173:475–82. - PubMed
-
- Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. - PubMed
-
- Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med. 2007;58:157–70. - PubMed
-
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–73. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
