

Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3
- PMID: 21643018
- PMCID: PMC3347888
- DOI: 10.1038/onc.2011.185
Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3
Abstract
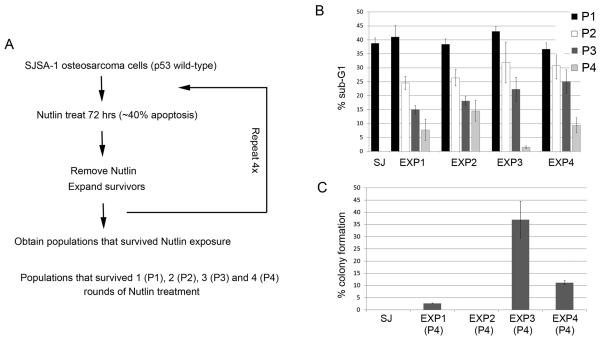
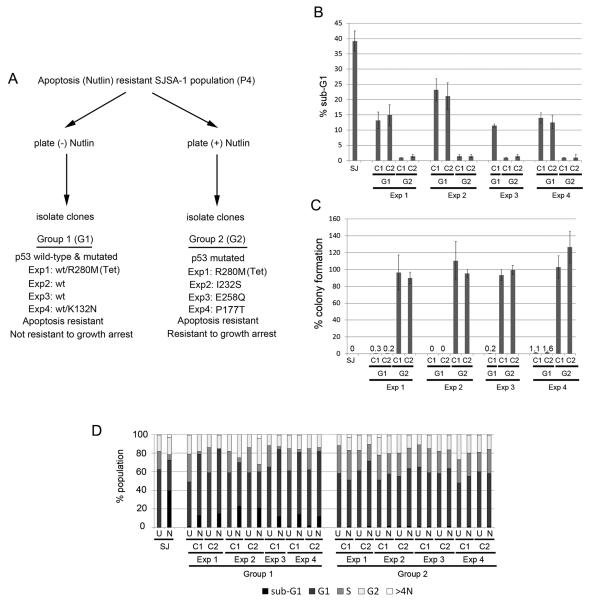
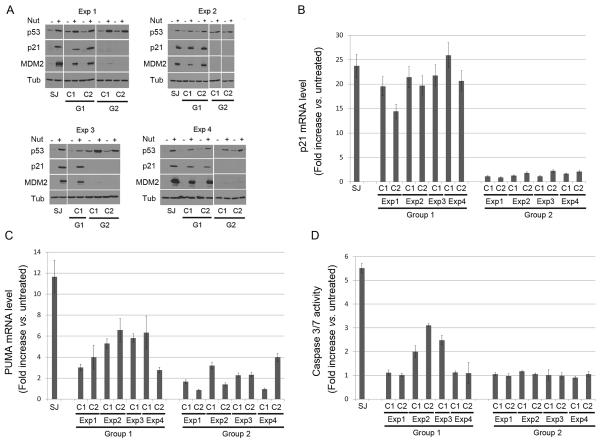
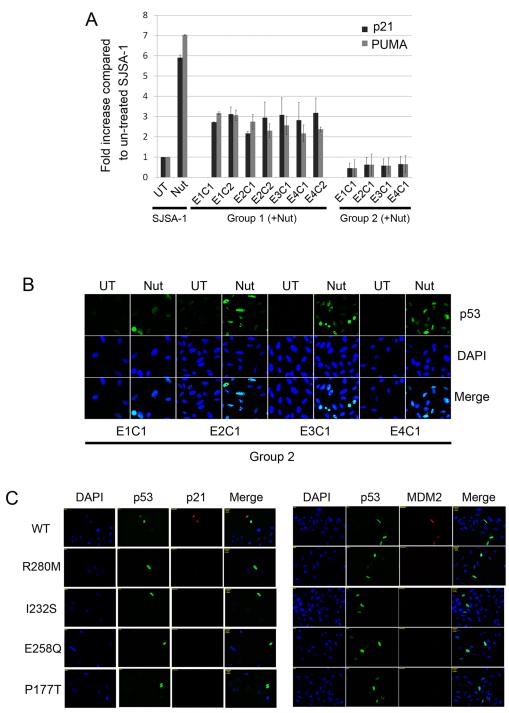
Wild-type p53 is a stress-responsive tumor suppressor and potent growth inhibitor. Genotoxic stresses (for example, ionizing and ultraviolet radiation or chemotherapeutic drug treatment) can activate p53, but also induce mutations in the P53 gene, and thus select for p53-mutated cells. Nutlin-3a (Nutlin) is pre-clinical drug that activates p53 in a non-genotoxic manner. Nutlin occupies the p53-binding pocket of murine double minute 2 (MDM2), activating p53 by blocking the p53-MDM2 interaction. Because Nutlin neither binds p53 directly nor introduces DNA damage, we hypothesized Nutlin would not induce P53 mutations, and, therefore, not select for p53-mutated cells. To test this, populations of SJSA-1 (p53 wild-type) cancer cells were expanded that survived repeated Nutlin exposures, and individual clones were isolated. Group 1 clones were resistant to Nutlin-induced apoptosis, but still underwent growth arrest. Surprisingly, while some Group 1 clones retained wild-type p53, others acquired a heterozygous p53 mutation. Apoptosis resistance in Group 1 clones was associated with decreased PUMA induction and decreased caspase 3/7 activation. Group 2 clones were resistant to both apoptosis and growth arrest induced by Nutlin. Group 2 clones had acquired mutations in the p53-DNA-binding domain and expressed only mutant p53s that were induced by Nutlin treatment, but were unable to bind the P21 and PUMA gene promoters, and unable to activate transcription. These results demonstrate that non-genotoxic p53 activation (for example, by Nutlin treatment) can lead to the acquisition of somatic mutations in p53 and select for p53-mutated cells. These findings have implications for the potential clinical use of Nutlin and other small molecule MDM2 antagonists.
Figures
References
-
- Ashkenazi A. Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev. 2008;19:325–31. - PubMed
-
- Blagosklonny MV. Oncogenic resistance to growth-limiting conditions. Nat Rev Cancer. 2002;2:221–5. - PubMed
-
- Blagosklonny MV. Carcinogenesis, cancer therapy and chemoprevention. Cell Death Differ. 2005;12:592–602. - PubMed
-
- Brown L, Boswell S, Raj L, Lee SW. Transcriptional targets of p53 that regulate cellular proliferation. Crit Rev Eukaryot Gene Expr. 2007;17:73–85. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
