

RNA-seq: technical variability and sampling
- PMID: 21645359
- PMCID: PMC3141664
- DOI: 10.1186/1471-2164-12-293
RNA-seq: technical variability and sampling
Abstract
Background: RNA-seq is revolutionizing the way we study transcriptomes. mRNA can be surveyed without prior knowledge of gene transcripts. Alternative splicing of transcript isoforms and the identification of previously unknown exons are being reported. Initial reports of differences in exon usage, and splicing between samples as well as quantitative differences among samples are beginning to surface. Biological variation has been reported to be larger than technical variation. In addition, technical variation has been reported to be in line with expectations due to random sampling. However, strategies for dealing with technical variation will differ depending on the magnitude. The size of technical variance, and the role of sampling are examined in this manuscript.
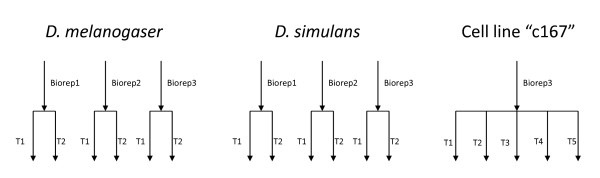
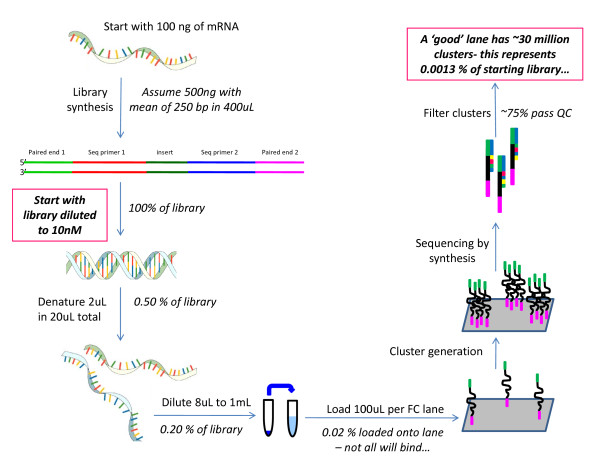
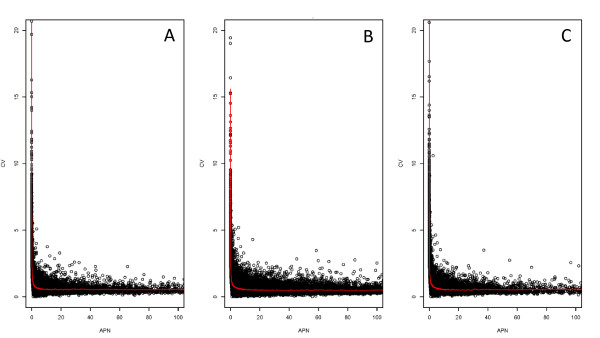
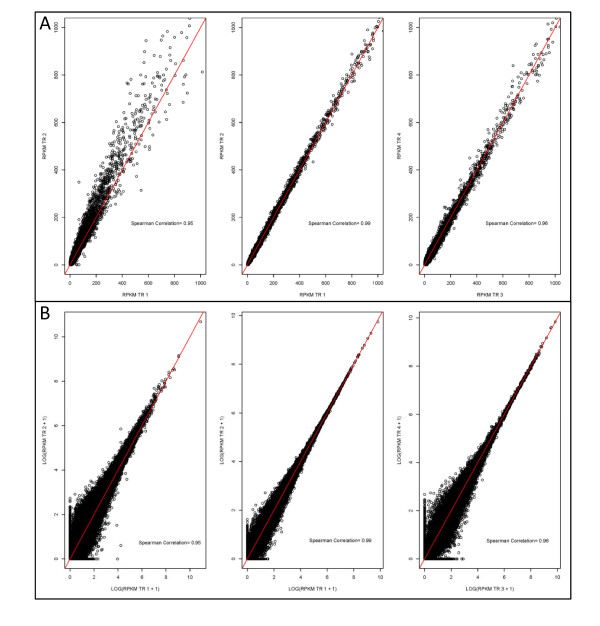
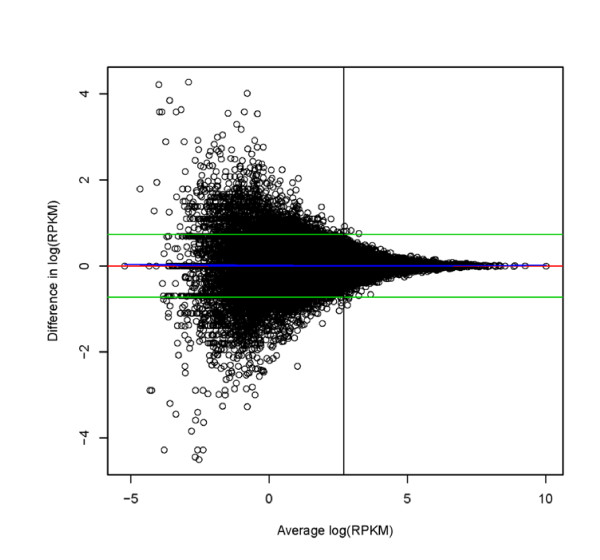
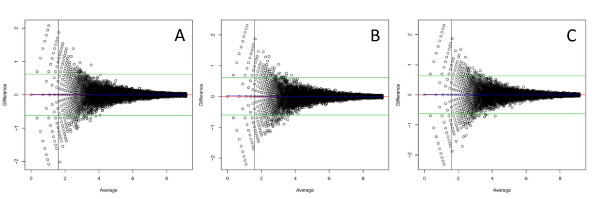
Results: In this study three independent Solexa/Illumina experiments containing technical replicates are analyzed. When coverage is low, large disagreements between technical replicates are apparent. Exon detection between technical replicates is highly variable when the coverage is less than 5 reads per nucleotide and estimates of gene expression are more likely to disagree when coverage is low. Although large disagreements in the estimates of expression are observed at all levels of coverage.
Conclusions: Technical variability is too high to ignore. Technical variability results in inconsistent detection of exons at low levels of coverage. Further, the estimate of the relative abundance of a transcript can substantially disagree, even when coverage levels are high. This may be due to the low sampling fraction and if so, it will persist as an issue needing to be addressed in experimental design even as the next wave of technology produces larger numbers of reads. We provide practical recommendations for dealing with the technical variability, without dramatic cost increases.
Figures
References
-
- Asmann Y, Klee E, Thompson EA, Perez E, Middha S, Oberg A, Therneau T, Smith D, Poland G, Wieben E, Kocher JP. 3' tag digital gene expression profiling of human brain and universal reference RNA using Illumina Genome Analyzer. BMC Genomics. 2009;10:531. doi: 10.1186/1471-2164-10-531. - DOI - PMC - PubMed
-
- Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, Schmidt D, O'Keefe S, Haas S, Vingron M, Lehrach H, Yaspo ML. A Global View of Gene Activity and Alternative Splicing by Deep Sequencing of the Human Transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
