

Systematic bias in high-throughput sequencing data and its correction by BEADS
- PMID: 21646344
- PMCID: PMC3159482
- DOI: 10.1093/nar/gkr425
Systematic bias in high-throughput sequencing data and its correction by BEADS
Abstract
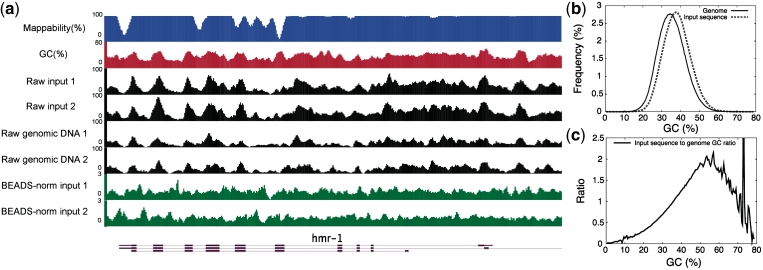
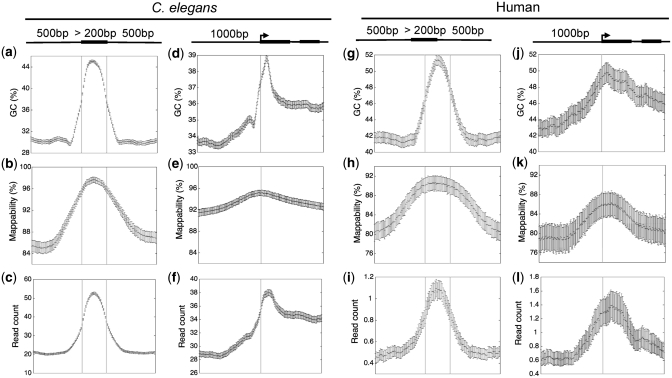
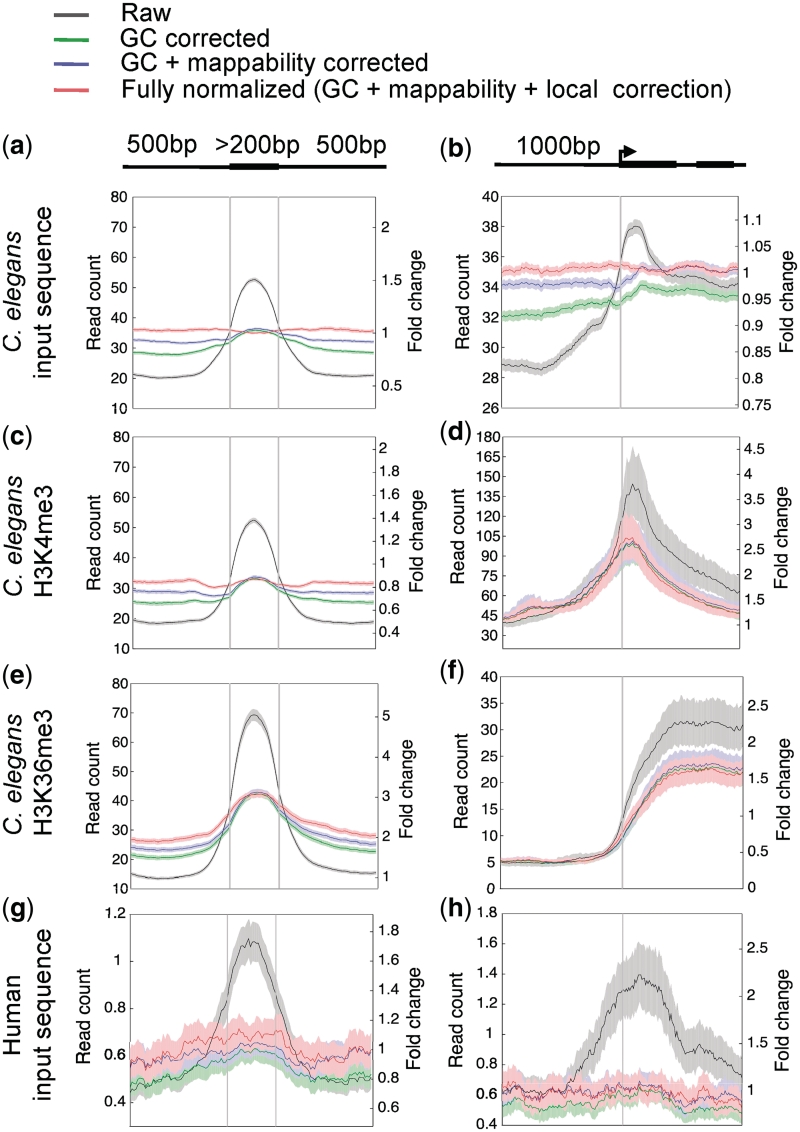
Genomic sequences obtained through high-throughput sequencing are not uniformly distributed across the genome. For example, sequencing data of total genomic DNA show significant, yet unexpected enrichments on promoters and exons. This systematic bias is a particular problem for techniques such as chromatin immunoprecipitation, where the signal for a target factor is plotted across genomic features. We have focused on data obtained from Illumina's Genome Analyser platform, where at least three factors contribute to sequence bias: GC content, mappability of sequencing reads, and regional biases that might be generated by local structure. We show that relying on input control as a normalizer is not generally appropriate due to sample to sample variation in bias. To correct sequence bias, we present BEADS (bias elimination algorithm for deep sequencing), a simple three-step normalization scheme that successfully unmasks real binding patterns in ChIP-seq data. We suggest that this procedure be done routinely prior to data interpretation and downstream analyses.
Figures
References
-
- Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
