

Regulation of VEGF-induced endothelial cell migration by mitochondrial reactive oxygen species
- PMID: 21653897
- PMCID: PMC3174570
- DOI: 10.1152/ajpcell.00322.2010
Regulation of VEGF-induced endothelial cell migration by mitochondrial reactive oxygen species
Abstract
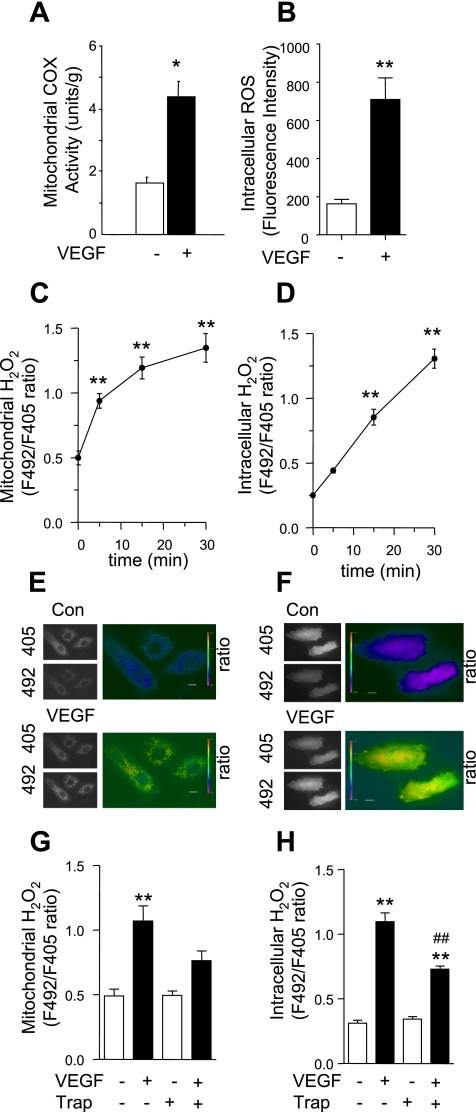
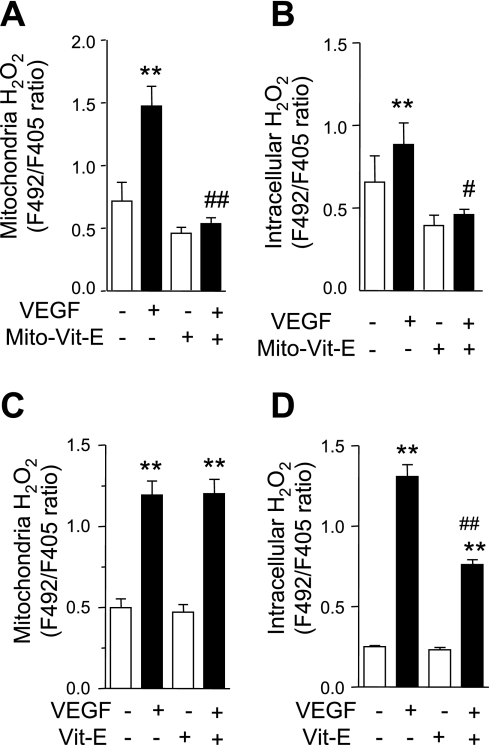
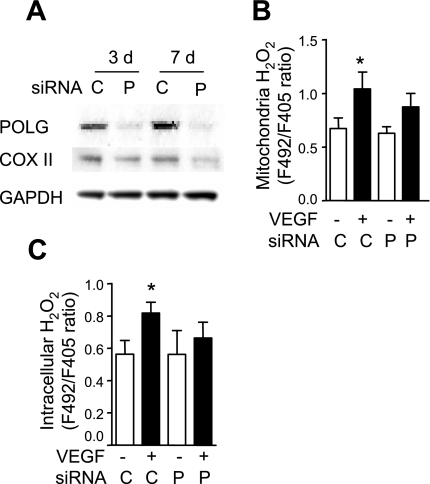
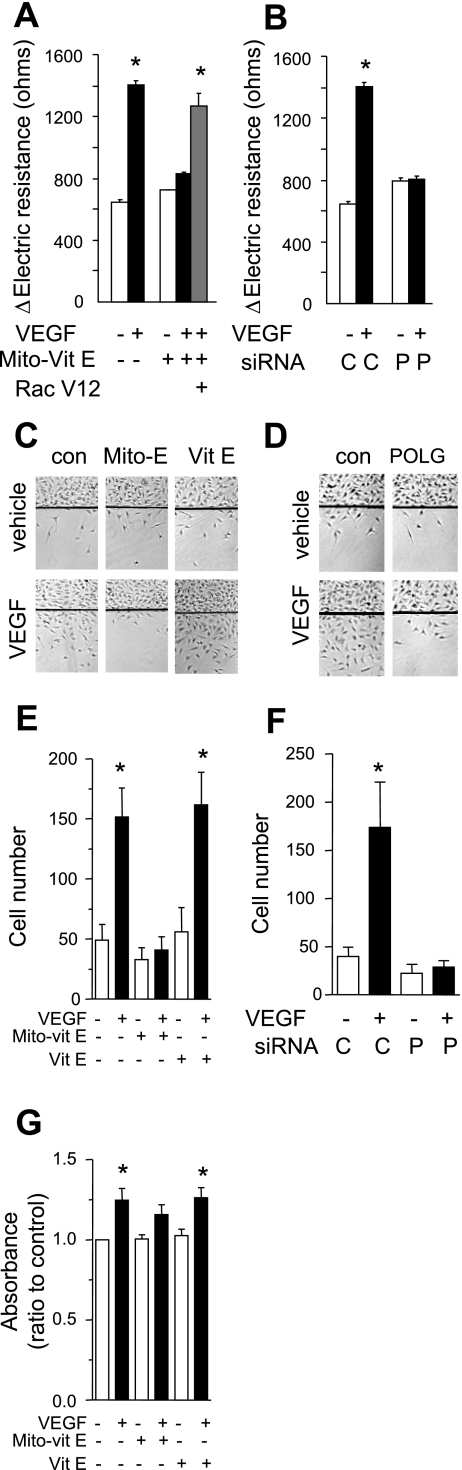
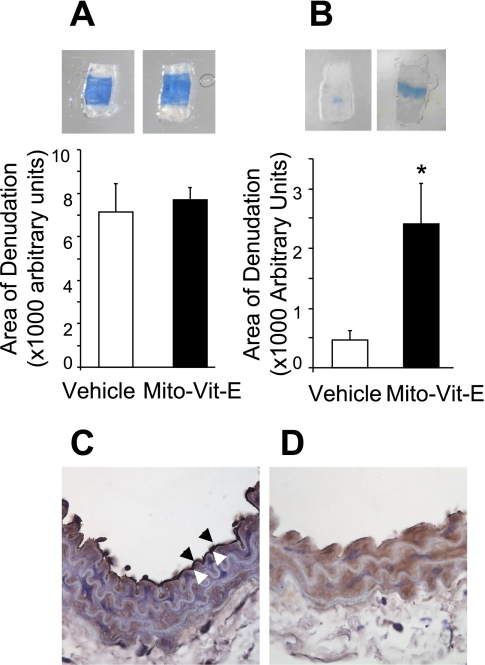
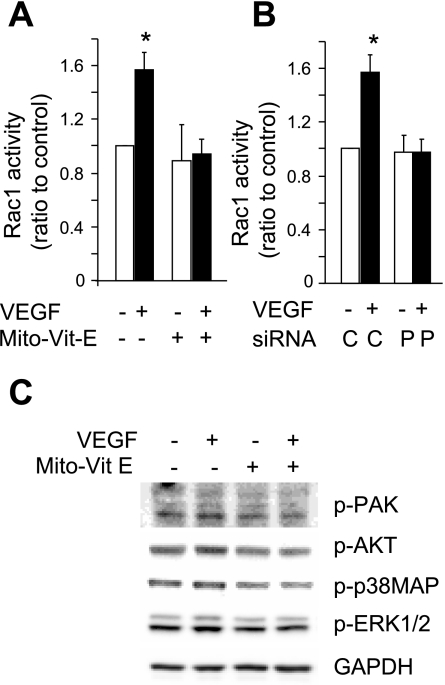
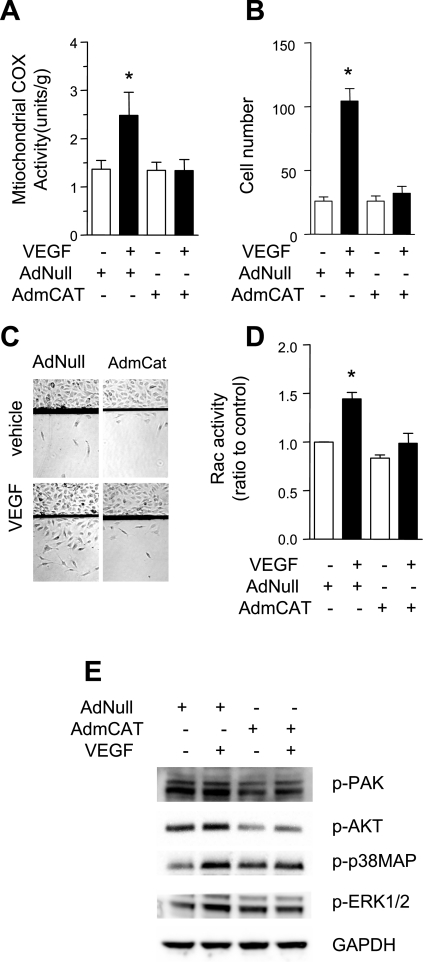
Endothelial migration is a crucial aspect of a variety of physiologic and pathologic conditions including atherosclerosis and vascular repair. Reactive oxygen species (ROS) function as second messengers during endothelial migration. Multiple intracellular sources of ROS are regulated by cellular context, external stimulus, and the microenvironment. However, the predominant source of ROS during endothelial cell (EC) migration and the mechanisms by which ROS regulate cell migration are incompletely understood. In this study, we tested the hypothesis that mitochondria-derived ROS (mtROS) regulate EC migration. In cultured human umbilical vein endothelial cells, VEGF increased mitochondrial metabolism, promoted mtROS production, and induced cell migration. Either the targeted mitochondrial delivery of the antioxidant, vitamin E (Mito-Vit-E), or the depletion of mitochondrial DNA abrogated VEGF-mediated mtROS production. Overexpression of mitochondrial catalase also inhibited VEGF-induced mitochondrial metabolism, Rac activation, and cell migration. Furthermore, these interventions suppressed VEGF-stimulated EC migration and blocked Rac1 activation in endothelial cells. Constitutively active Rac1 reversed Mito-Vit-E-induced inhibition of EC migration. Mito-Vit-E also attenuated carotid artery reendothelialization in vivo. These results provide strong evidence that mtROS regulate EC migration through Rac-1.
Figures
References
-
- Abid MR, Spokes KC, Shih SC, Aird WC. NADPH oxidase activity selectively modulates vascular endothelial growth factor signaling pathways. J Biol Chem 282: 35373–35385, 2007 - PubMed
-
- Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J 19: 1088–1095, 2005 - PubMed
-
- Ahmad IM, Aykin-Burns N, Sim JE, Walsh SA, Higashikubo R, Buettner GR, Venkataraman S, Mackey MA, Flanagan SW, Oberley LW, Spitz DR. Mitochondrial O2*− and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J Biol Chem 280: 4254–4263, 2005 - PubMed
-
- Bai J, Rodriguez AM, Melendez JA, Cederbaum AI. Overexpression of catalase in cytosolic or mitochondrial compartment protects HepG2 cells against oxidative injury. J Biol Chem 274: 26217–26224, 1999 - PubMed
-
- Burridge K, Wennerberg K. Rho and Rac take center stage. Cell 116: 167–179, 2004 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous