

RNA sequencing reveals two major classes of gene expression levels in metazoan cells
- PMID: 21654674
- PMCID: PMC3159973
- DOI: 10.1038/msb.2011.28
RNA sequencing reveals two major classes of gene expression levels in metazoan cells
Abstract
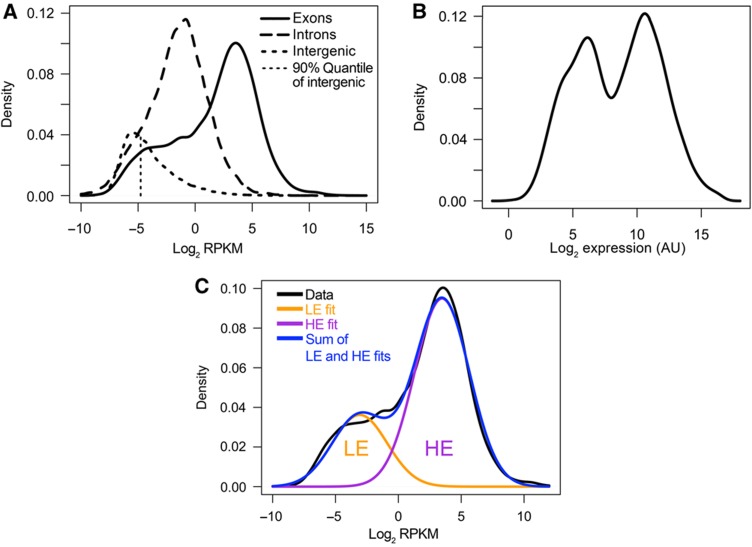
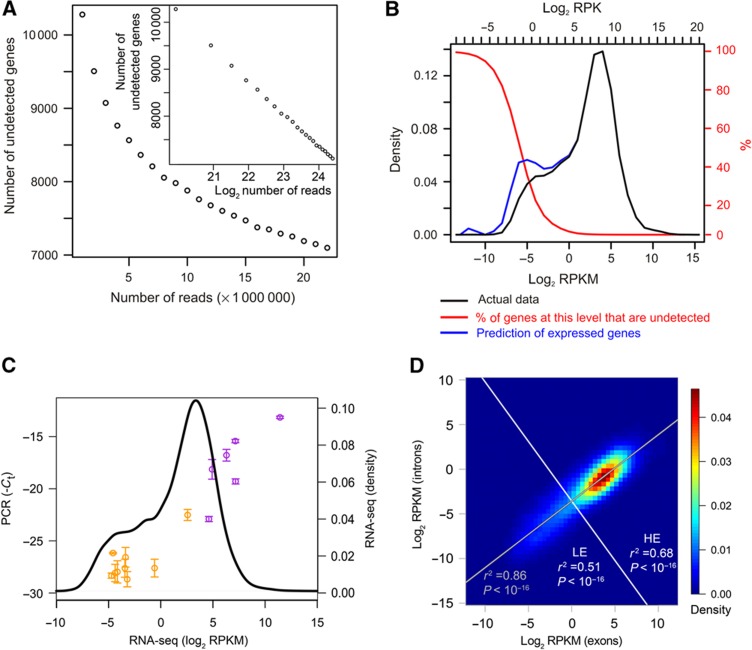
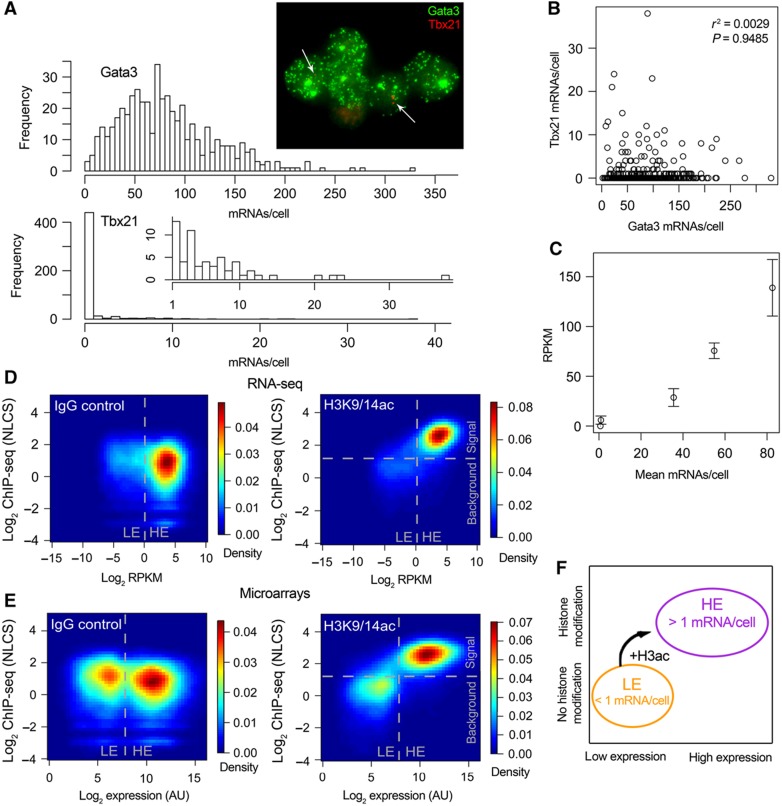
The expression level of a gene is often used as a proxy for determining whether the protein or RNA product is functional in a cell or tissue. Therefore, it is of fundamental importance to understand the global distribution of gene expression levels, and to be able to interpret it mechanistically and functionally. Here we use RNA sequencing (RNA-seq) of mouse Th2 cells, coupled with a range of other techniques, to show that all genes can be separated, based on their expression abundance, into two distinct groups: one group comprised of lowly expressed and putatively non-functional mRNAs, and the other of highly expressed mRNAs with active chromatin marks at their promoters. These observations are confirmed in many other microarray and RNA-seq data sets of metazoan cell types.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Akaike H (1974) New look at statistical-model identification. Ieee T Automat Contr Ac 19: 716–723
-
- Casella G, Berger RL (2001) Statistical Inference, 2nd edn. Pacific Grove, CA, USA: Duxbury Press
-
- Chintapalli VR, Wang J, Dow JA (2007) Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet 39: 715–720 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
