

Mutations in PRDM5 in brittle cornea syndrome identify a pathway regulating extracellular matrix development and maintenance
- PMID: 21664999
- PMCID: PMC3113239
- DOI: 10.1016/j.ajhg.2011.05.007
Mutations in PRDM5 in brittle cornea syndrome identify a pathway regulating extracellular matrix development and maintenance
Erratum in
- Am J Hum Genet. 2011 Aug 12;89(2):346
Abstract
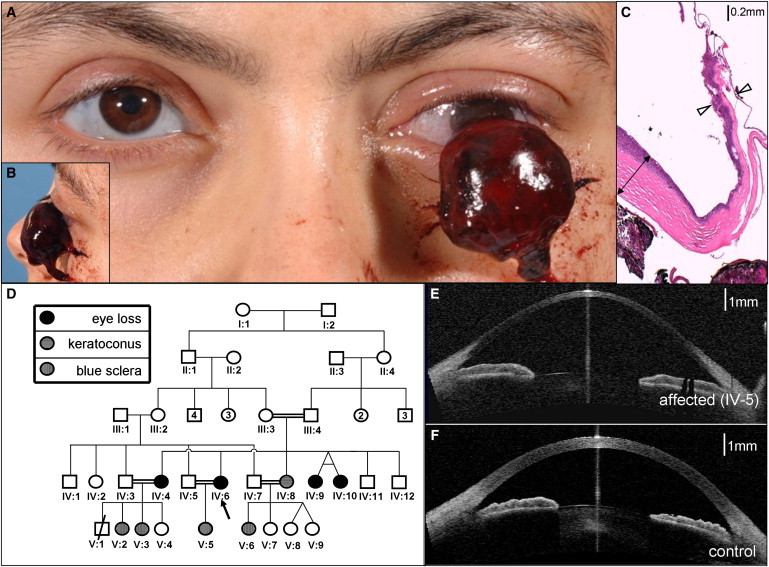
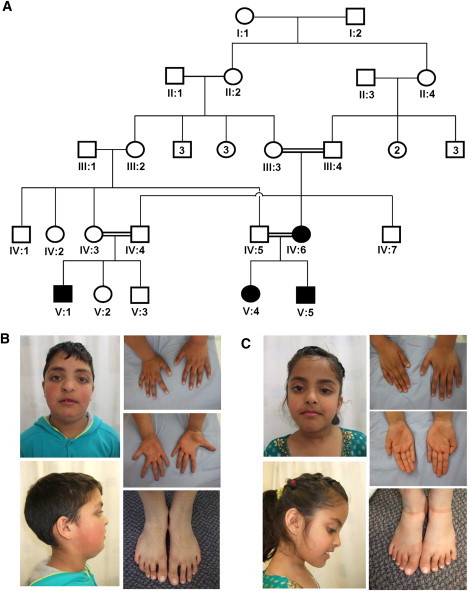
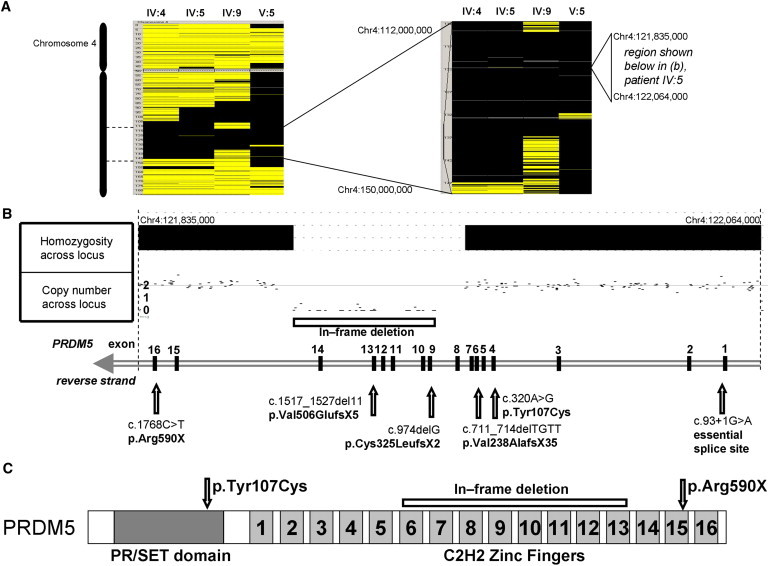
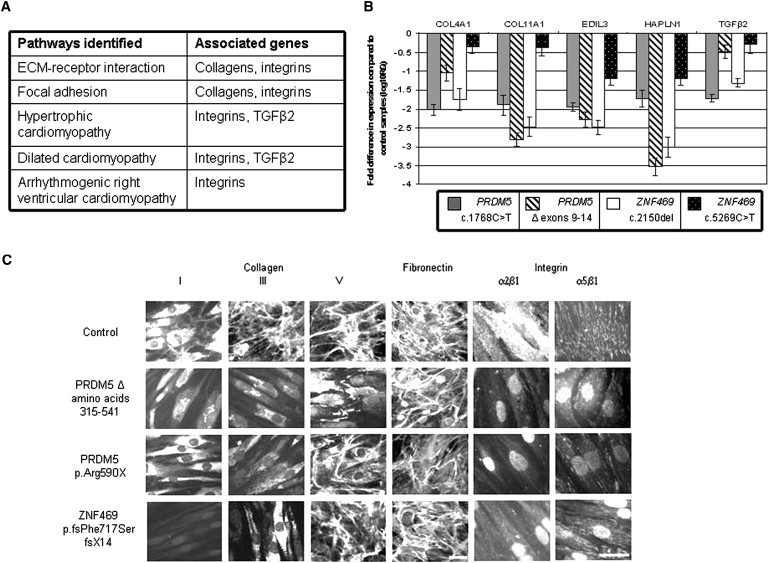
Extreme corneal fragility and thinning, which have a high risk of catastrophic spontaneous rupture, are the cardinal features of brittle cornea syndrome (BCS), an autosomal-recessive generalized connective tissue disorder. Enucleation is frequently the only management option for this condition, resulting in blindness and psychosocial distress. Even when the cornea remains grossly intact, visual function could also be impaired by a high degree of myopia and keratoconus. Deafness is another common feature and results in combined sensory deprivation. Using autozygosity mapping, we identified mutations in PRDM5 in families with BCS. We demonstrate that regulation of expression of extracellular matrix components, particularly fibrillar collagens, by PRDM5 is a key molecular mechanism that underlies corneal fragility in BCS and controls normal corneal development and maintenance. ZNF469, encoding a zinc finger protein of hitherto undefined function, has been identified as a quantitative trait locus for central corneal thickness, and mutations in this gene have been demonstrated in Tunisian Jewish and Palestinian kindreds with BCS. We show that ZNF469 and PRDM5, two genes that when mutated cause BCS, participate in the same regulatory pathway.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Al-Hussain H., Zeisberger S.M., Huber P.R., Giunta C., Steinmann B. Brittle cornea syndrome and its delineation from the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VI): Report on 23 patients and review of the literature. Am. J. Med. Genet. A. 2004;124A:28–34. - PubMed
-
- Izquierdo L., Jr., Mannis M.J., Marsh P.B., Yang S.P., McCarthy J.M. Bilateral spontaneous corneal rupture in brittle cornea syndrome: A case report. Cornea. 1999;18:621–624. - PubMed
-
- Christensen A.E., Knappskog P.M., Midtbø M., Gjesdal C.G., Mengel-From J., Morling N., Rødahl E., Boman H. Brittle cornea syndrome associated with a missense mutation in the zinc-finger 469 gene. Invest. Ophthalmol. Vis. Sci. 2010;51:47–52. - PubMed
-
- Khan A.O., Aldahmesh M.A., Mohamed J.N., Alkuraya F.S. Blue sclera with and without corneal fragility (brittle cornea syndrome) in a consanguineous family harboring ZNF469 mutation (p.E1392X) Arch. Ophthalmol. 2010;128:1376–1379. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
