

Cactus: Algorithms for genome multiple sequence alignment
- PMID: 21665927
- PMCID: PMC3166836
- DOI: 10.1101/gr.123356.111
Cactus: Algorithms for genome multiple sequence alignment
Abstract
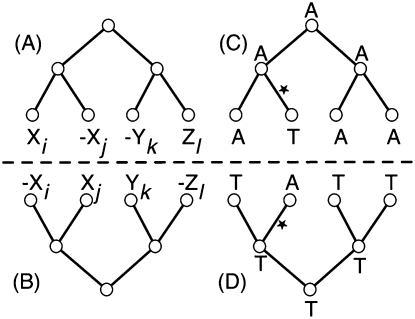
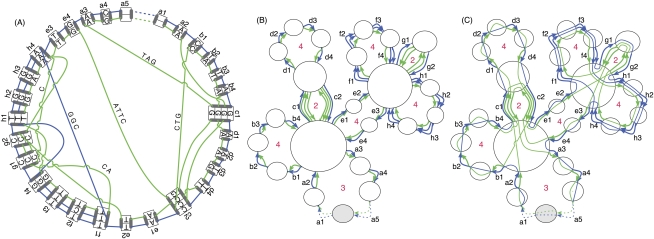
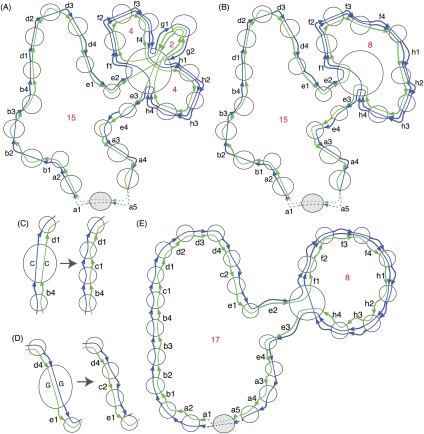
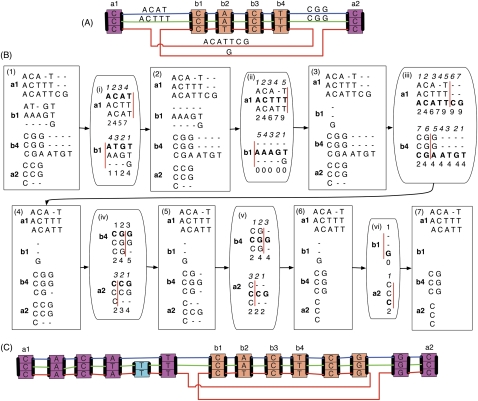
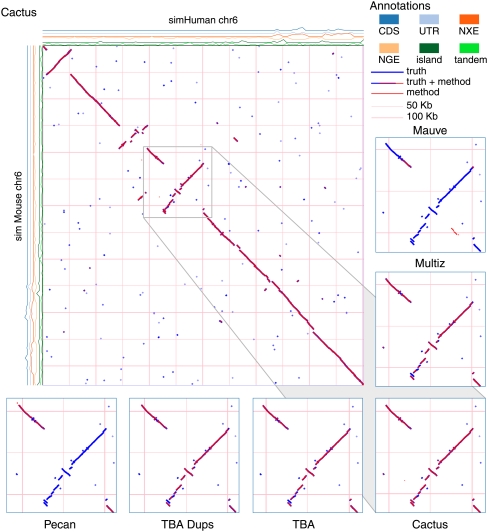
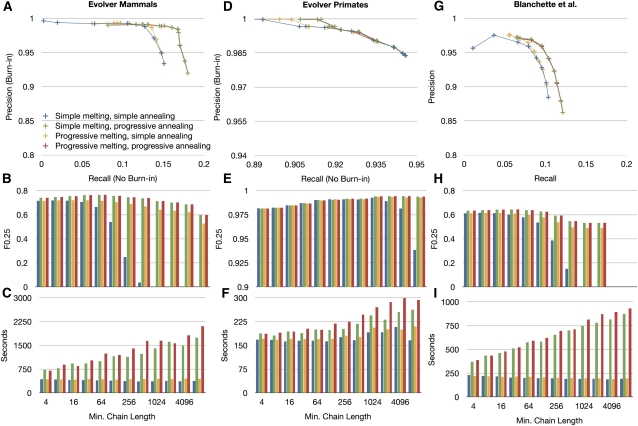
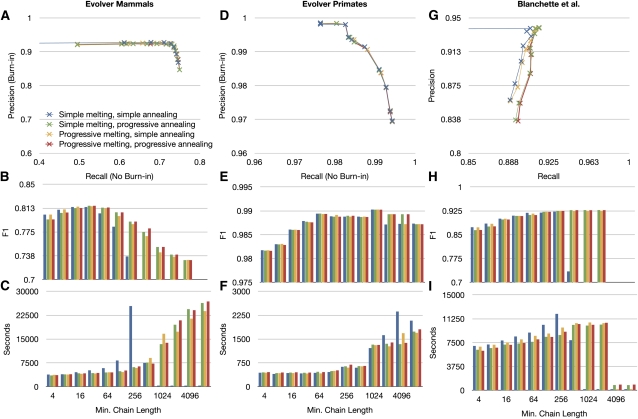
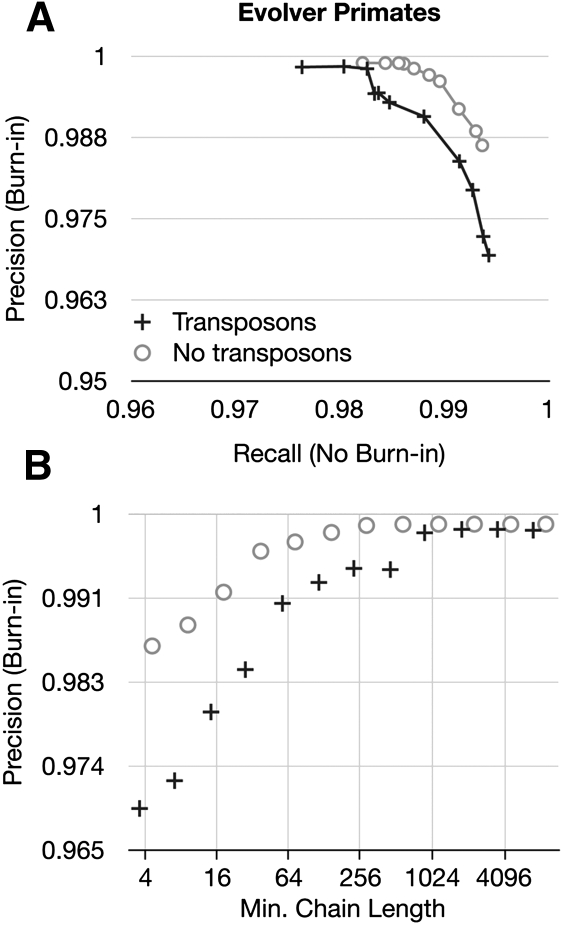
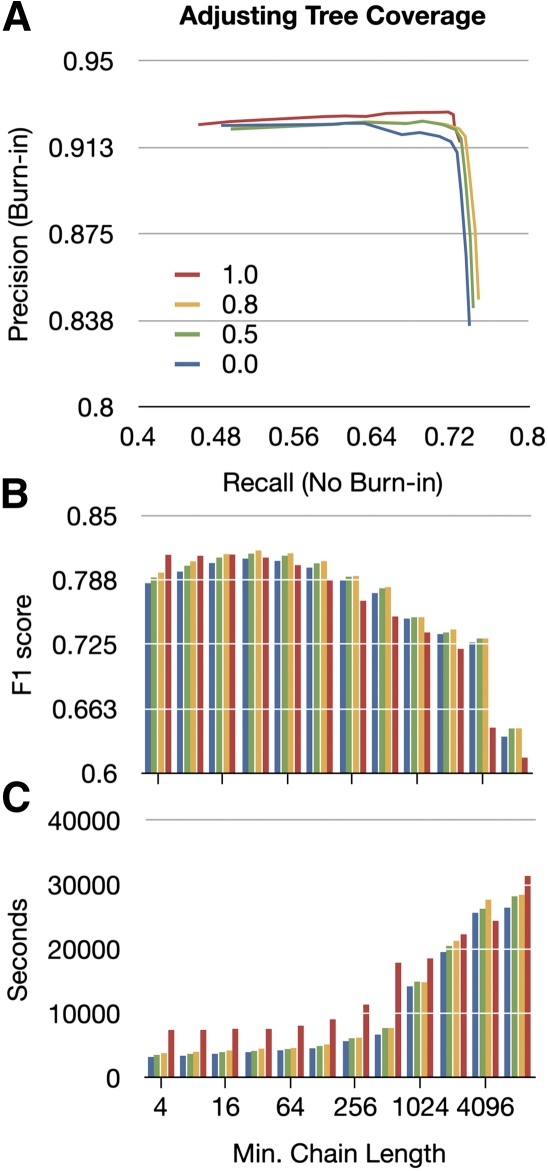
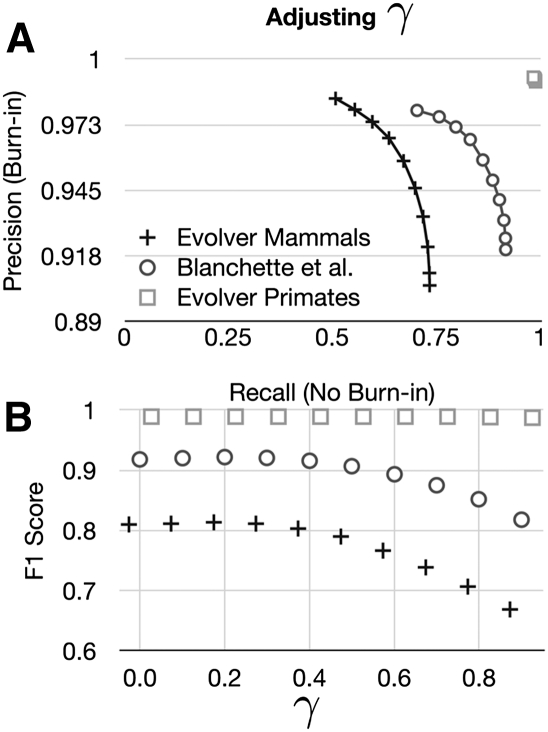
Much attention has been given to the problem of creating reliable multiple sequence alignments in a model incorporating substitutions, insertions, and deletions. Far less attention has been paid to the problem of optimizing alignments in the presence of more general rearrangement and copy number variation. Using Cactus graphs, recently introduced for representing sequence alignments, we describe two complementary algorithms for creating genomic alignments. We have implemented these algorithms in the new "Cactus" alignment program. We test Cactus using the Evolver genome evolution simulator, a comprehensive new tool for simulation, and show using these and existing simulations that Cactus significantly outperforms all of its peers. Finally, we make an empirical assessment of Cactus's ability to properly align genes and find interesting cases of intra-gene duplication within the primates.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources