

Patterns and mechanisms of ancestral histone protein inheritance in budding yeast
- PMID: 21666805
- PMCID: PMC3110181
- DOI: 10.1371/journal.pbio.1001075
Patterns and mechanisms of ancestral histone protein inheritance in budding yeast
Abstract
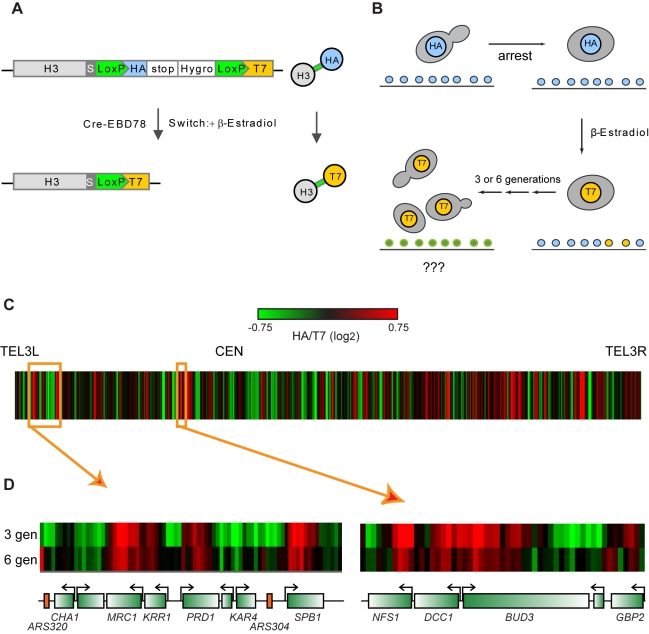
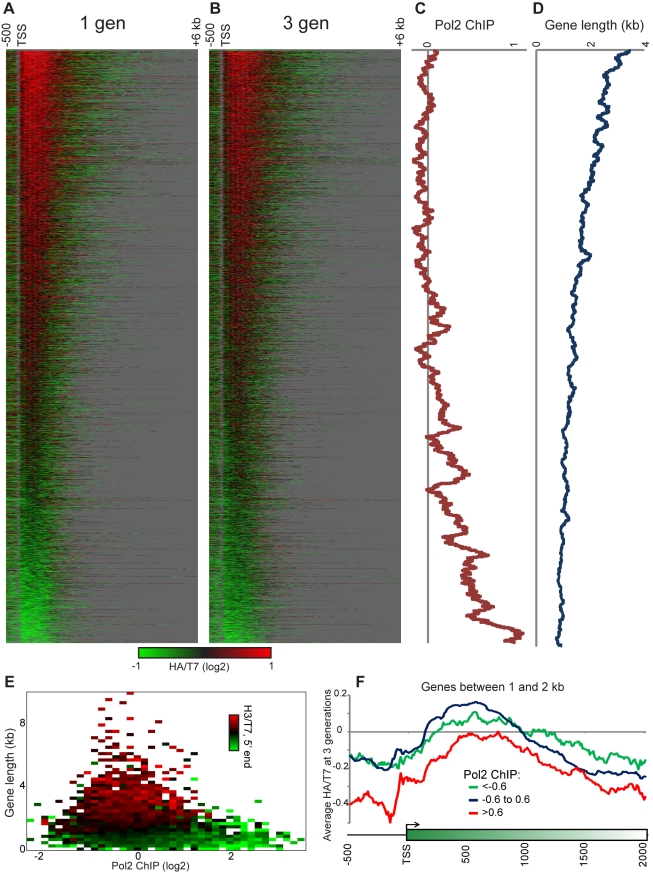
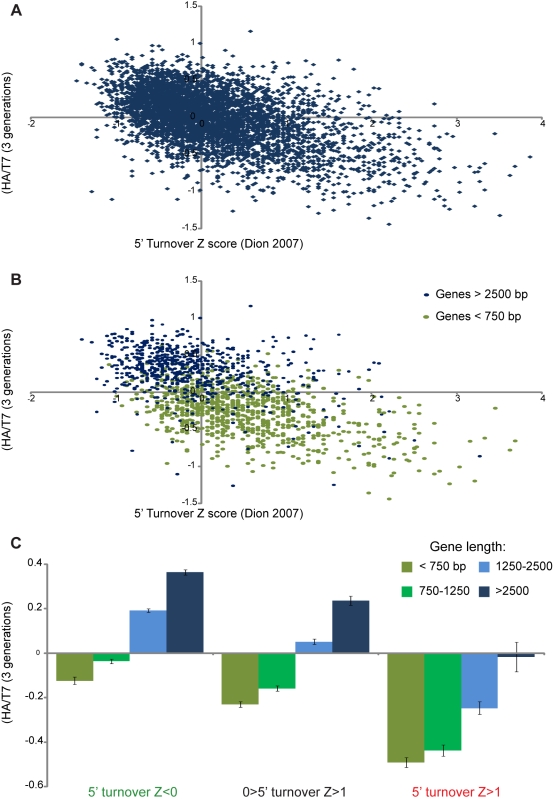
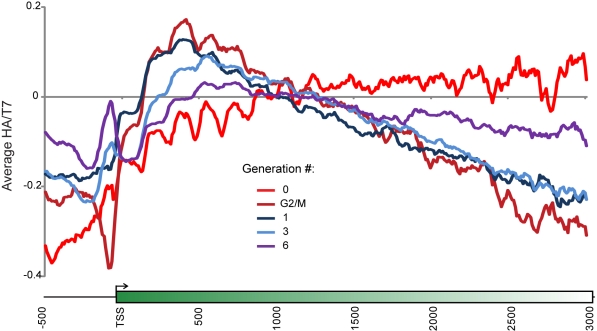
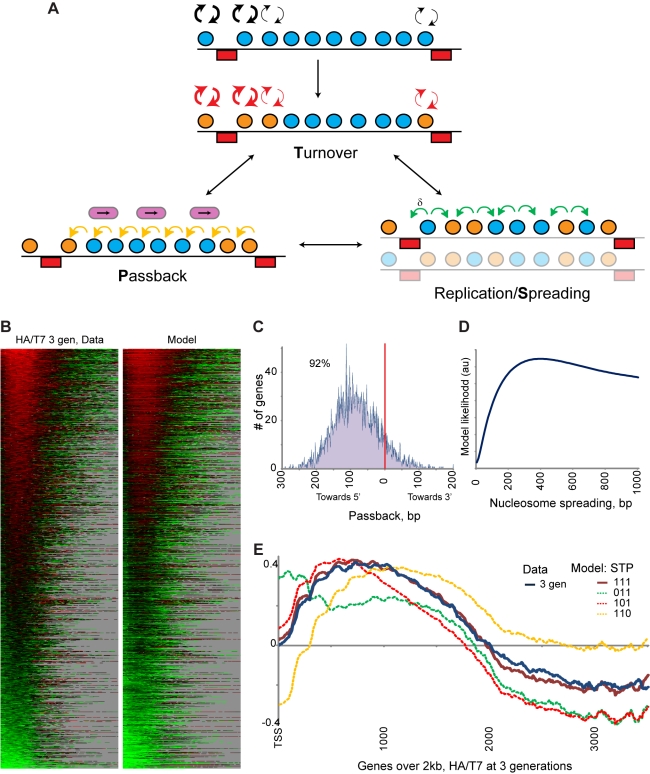
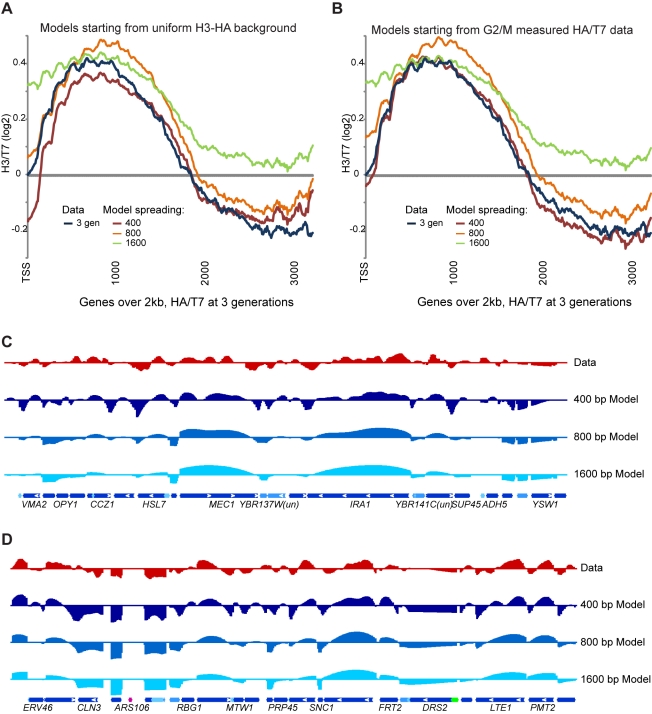
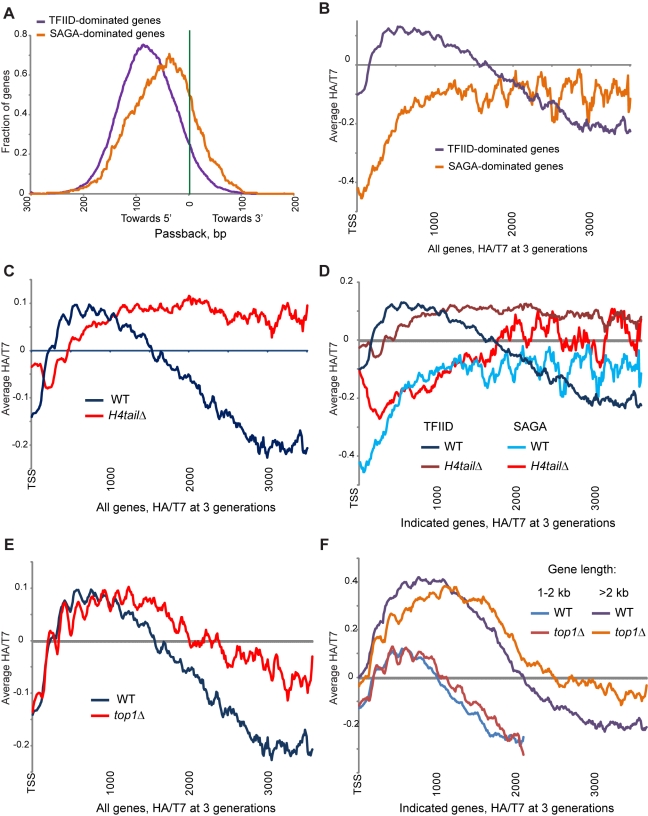
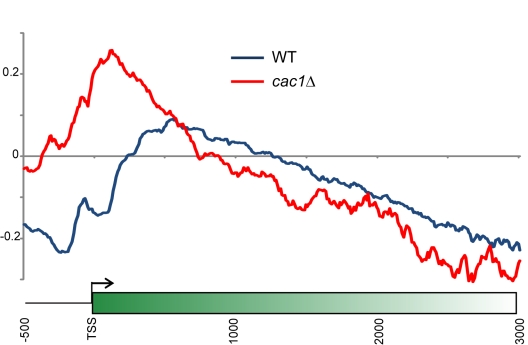
Replicating chromatin involves disruption of histone-DNA contacts and subsequent reassembly of maternal histones on the new daughter genomes. In bulk, maternal histones are randomly segregated to the two daughters, but little is known about the fine details of this process: do maternal histones re-assemble at preferred locations or close to their original loci? Here, we use a recently developed method for swapping epitope tags to measure the disposition of ancestral histone H3 across the yeast genome over six generations. We find that ancestral H3 is preferentially retained at the 5' ends of most genes, with strongest retention at long, poorly transcribed genes. We recapitulate these observations with a quantitative model in which the majority of maternal histones are reincorporated within 400 bp of their pre-replication locus during replication, with replication-independent replacement and transcription-related retrograde nucleosome movement shaping the resulting distributions of ancestral histones. We find a key role for Topoisomerase I in retrograde histone movement during transcription, and we find that loss of Chromatin Assembly Factor-1 affects replication-independent turnover. Together, these results show that specific loci are enriched for histone proteins first synthesized several generations beforehand, and that maternal histones re-associate close to their original locations on daughter genomes after replication. Our findings further suggest that accumulation of ancestral histones could play a role in shaping histone modification patterns.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Comment in
-
Histones are passed back to stay in place, more or less.PLoS Biol. 2011 Jun;9(6):e1001072. doi: 10.1371/journal.pbio.1001072. Epub 2011 Jun 7. PLoS Biol. 2011. PMID: 21666804 Free PMC article. No abstract available.
References
-
- Ptashne M. On the use of the word ‘epigenetic’. Curr Biol. 2007;17:R233–R236. - PubMed
-
- Ringrose L, Paro R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet. 2004;38:413–443. - PubMed
-
- Rusche L. N, Kirchmaier A. L, Rine J. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem. 2003;72:481–516. - PubMed
-
- Annunziato A. T. Split decision: what happens to nucleosomes during DNA replication? J Biol Chem. 2005;280:12065–12068. - PubMed
-
- Groth A. Replicating chromatin: a tale of histones. Biochem Cell Biol. 2009;87:51–63. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
