

Viral infection augments Nod1/2 signaling to potentiate lethality associated with secondary bacterial infections
- PMID: 21669398
- PMCID: PMC3125972
- DOI: 10.1016/j.chom.2011.05.006
Viral infection augments Nod1/2 signaling to potentiate lethality associated with secondary bacterial infections
Abstract
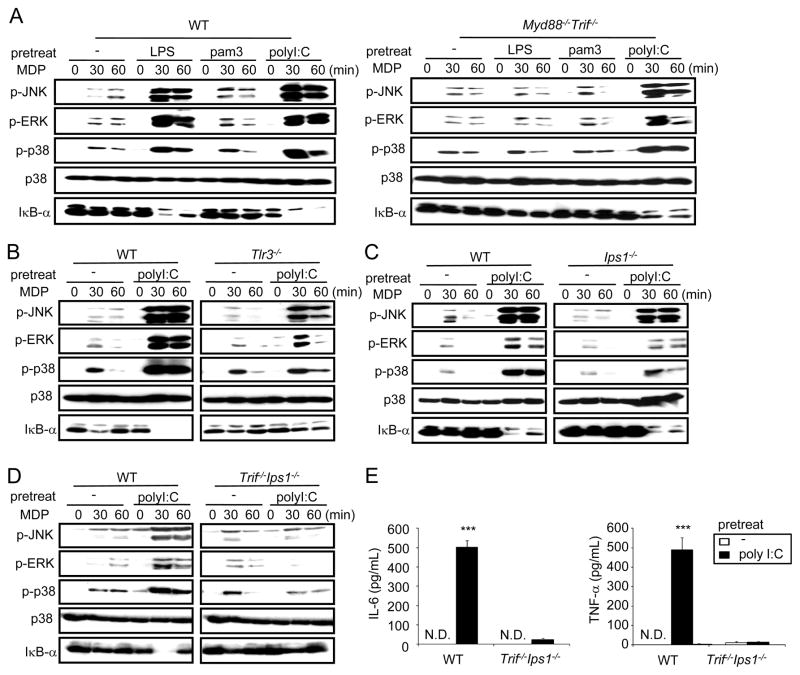
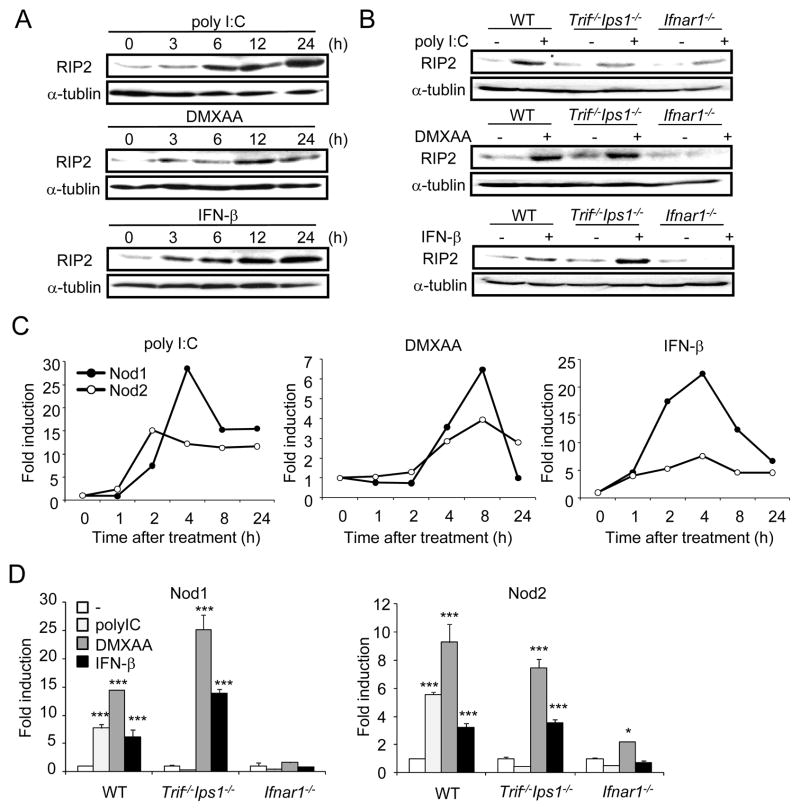
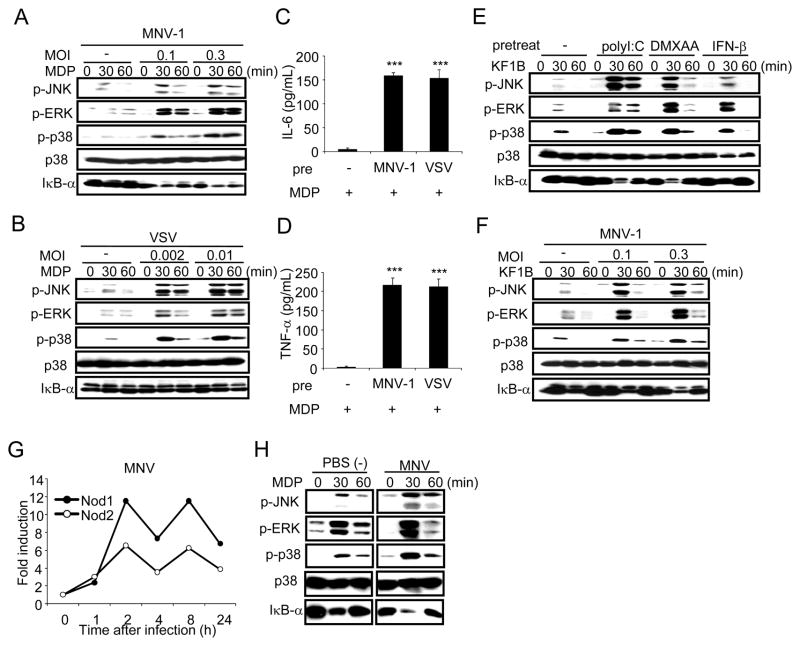
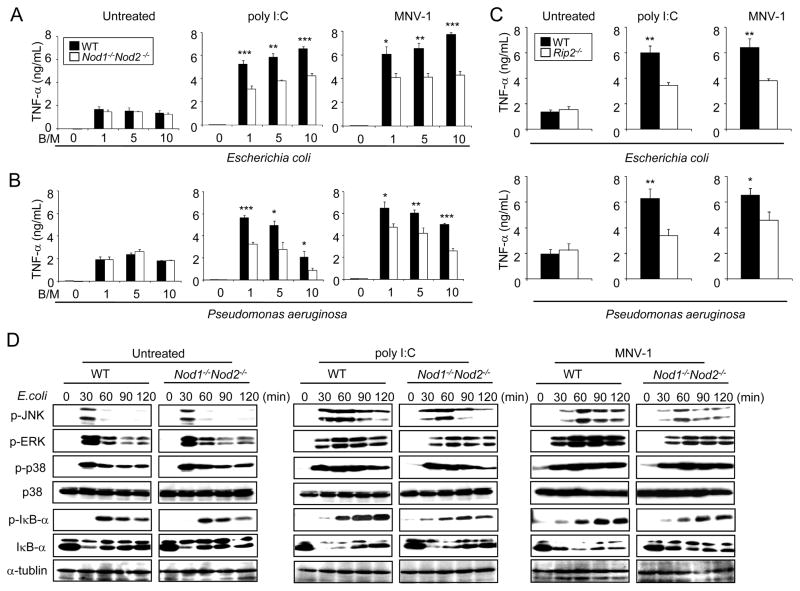
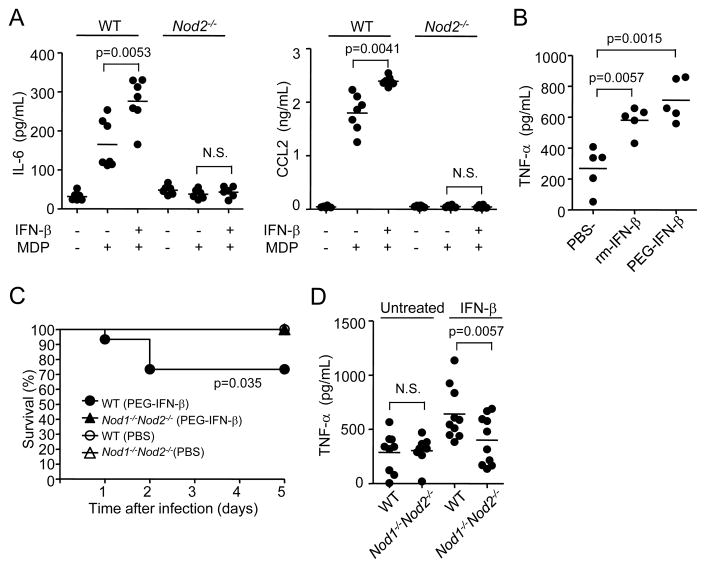
Secondary bacterial infection is a common sequela to viral infection and is associated with increased lethality and morbidity. However, the underlying mechanisms remain poorly understood. We show that the TLR3/MDA5 agonist poly I:C or viral infection dramatically augments signaling via the NLRs Nod1 and Nod2 and enhances the production of proinflammatory cytokines. Enhanced Nod1 and Nod2 signaling by poly I:C required the TLR3/MDA5 adaptors TRIF and IPS-1 and was mediated by type I IFNs. Mechanistically, poly I:C or IFN-β induced the expression of Nod1, Nod2, and the Nod-signaling adaptor Rip2. Systemic administration of poly I:C or IFN-β or infection with murine norovirus-1 promoted inflammation and lethality in mice superinfected with E. coli, which was independent of bacterial burden but attenuated in the absence of Nod1/Nod2 or Rip2. Thus, crosstalk between type I IFNs and Nod1/Nod2 signaling promotes bacterial recognition, but induces harmful effects in the virally infected host.
Copyright © 2011 Elsevier Inc. All rights reserved.
Conflict of interest statement
Darren P. Baker is employed by Biogen Idec. Biogen Idec provided unmodified and PEGylated murine IFN-β for these studies at no cost and did not provide funds for these studies, nor was Biogen Idec involved in the study design, data analysis or any decisions relating to manuscript preparation or submission.
Figures


Comment in
-
Immunology: virus and bacterium gang up on host.Nat Rev Microbiol. 2011 Aug 15;9(8):559. doi: 10.1038/nrmicro2628. Nat Rev Microbiol. 2011. PMID: 21760618 No abstract available.
References
-
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. - PubMed
-
- Beadling C, Slifka MK. How do viral infections predispose patients to bacterial infections? Curr Opin Infect Dis. 2004;17:185–191. - PubMed
-
- Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, Manogue KR, Cerami A, Bucala R. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365:756–759. - PubMed
-
- Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. 1985;229:869–871. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
