

A pilot study of bacterial genes with disrupted ORFs reveals a surprising profusion of protein sequence recoding mediated by ribosomal frameshifting and transcriptional realignment
- PMID: 21673094
- PMCID: PMC3199440
- DOI: 10.1093/molbev/msr155
A pilot study of bacterial genes with disrupted ORFs reveals a surprising profusion of protein sequence recoding mediated by ribosomal frameshifting and transcriptional realignment
Abstract
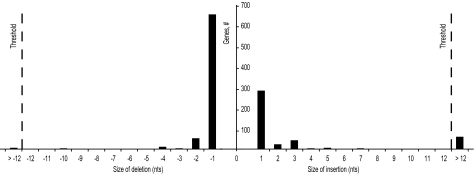
Bacterial genome annotations contain a number of coding sequences (CDSs) that, in spite of reading frame disruptions, encode a single continuous polypeptide. Such disruptions have different origins: sequencing errors, frameshift, or stop codon mutations, as well as instances of utilization of nontriplet decoding. We have extracted over 1,000 CDSs with annotated disruptions and found that about 75% of them can be clustered into 64 groups based on sequence similarity. Analysis of the clusters revealed deep phylogenetic conservation of open reading frame organization as well as the presence of conserved sequence patterns that indicate likely utilization of the nonstandard decoding mechanisms: programmed ribosomal frameshifting (PRF) and programmed transcriptional realignment (PTR). Further enrichment of these clusters with additional homologous nucleotide sequences revealed over 6,000 candidate genes utilizing PRF or PTR. Analysis of the patterns of conservation apparently associated with nontriplet decoding revealed the presence of both previously characterized frameshift-prone sequences and a few novel ones. Since the starting point of our analysis was a set of genes with already annotated disruptions, it is highly plausible that in this study, we have identified only a fraction of all bacterial genes that utilize PRF or PTR. In addition to the identification of a large number of recoded genes, a surprising observation is that nearly half of them are expressed via PTR-a mechanism that, in contrast to PRF, has not yet received substantial attention.
Figures







References
-
- Antonov I, Borodovsky M. Genetack: frameshift identification in protein-coding sequences by the Viterbi algorithm. J Bioinform Comput Biol. 2010;8:535–551. - PubMed
-
- Atkins JF, Baranov PV, Fayet O, et al. (13 co-authors) Overriding standard decoding: implications of recoding for ribosome function and enrichment of gene expression. Cold Spring Harb Symp Quant Biol. 2001;66:217–232. - PubMed
-
- Atkins JF, Gesteland RF, editors. Recoding: expansion of decoding rules enriches gene expression. New York: Springer; 2010.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
